Redox Biology|华中科技大学付琴教授团队提出靶向抑制PDE4D治疗心力衰竭的新兴策略
心力衰竭(HF)是全球主要死因之一,目前治疗手段有限。环磷酸腺苷(cAMP)在心脏的正常和病理信号传导中起着重要作用。PDE4是心脏中降解cAMP的主要磷酸二酯酶,PDE4的4B和4D亚型在心脏功能调节中的作用存在争议。PDE4B过表达对心脏重塑和HF有益,然而,PDE4D和PDE4抑制剂在心力衰竭中的作用仍不明确。2025年2月22日,华中科技大学同济医学院基础医学院付琴教授团队在Redox Biology (IF10.7) 发文“PDE4D inhibition ameliorates cardiac hypertrophy and heart failure by activating mitophagy”。该研究发现 PDE4D 在心力衰竭中表达增加,抑制PDE4D可通过激活线粒体自噬改善心肌肥厚和心力衰竭,为心力衰竭治疗提供新靶点和策略。
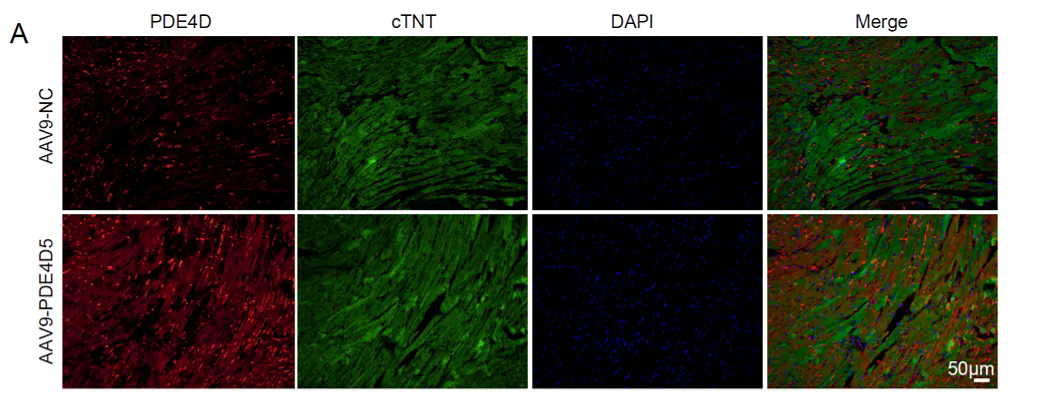
免疫荧光染色检测PDE4D在心肌细胞中特异性过表达
研究结果
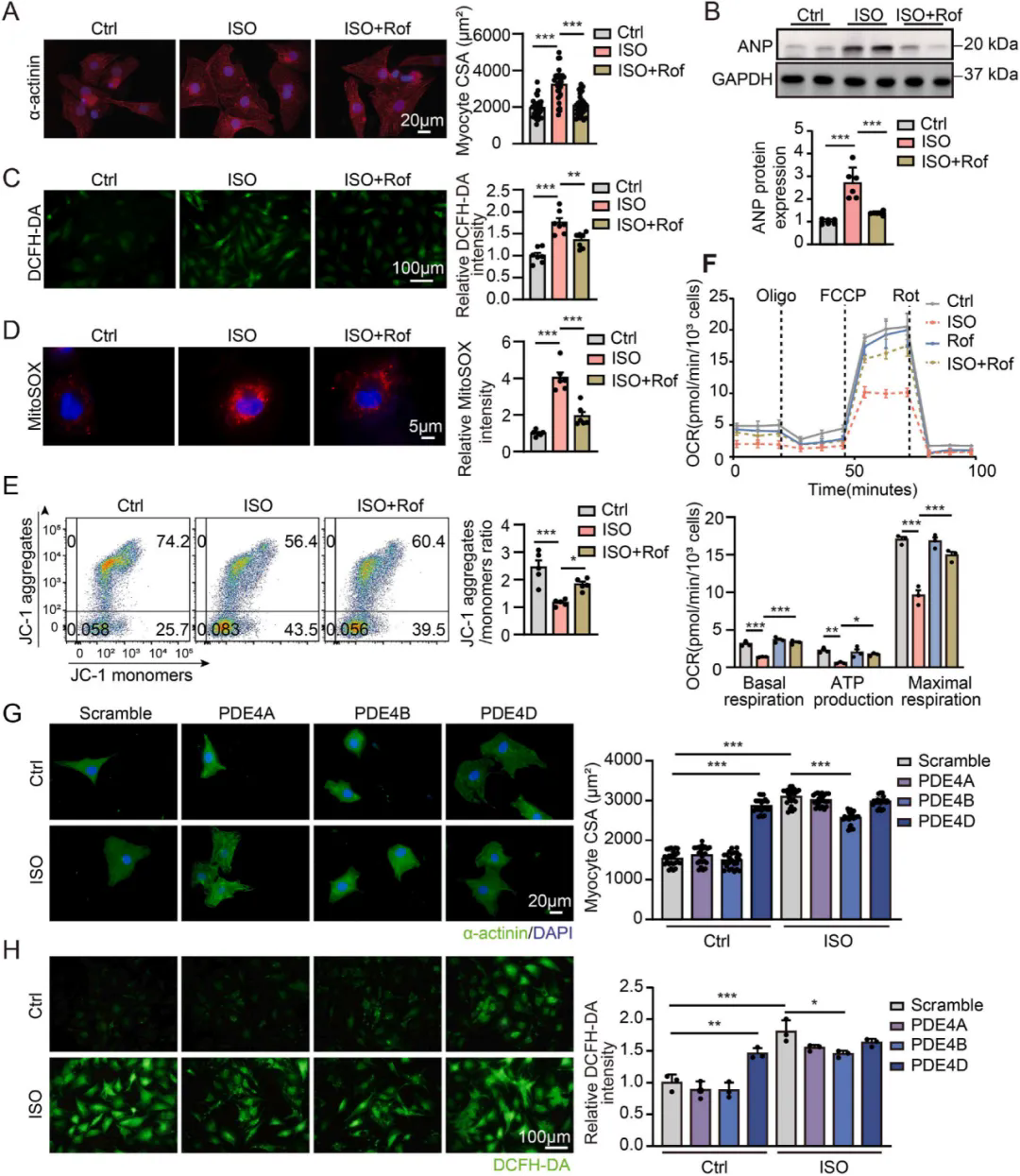
1、PDE4D诱导心肌细胞肥大和氧化应激
体内实验数据显示使用PDE4抑制剂罗氟司特可抑制异丙肾上腺素诱导的心肌肥大和心力衰竭,同时ISO处理使小鼠心脏PDE4D mRNA和蛋白水平上调,并降低PKA 活性;罗氟司特处理可抑制PDE4D的诱导,使PDE4活性、cAMP含量以及CREB和受磷蛋白的磷酸化水平恢复正常,且对PDE4A和PDE4B的表达无显著影响。体外实验进一步验证了PDE4抑制剂的心脏保护作用,通过对比PDE4D 和PDE4B对心肌细胞的不同影响,发现PDE4D可诱导心肌细胞肥大和氧化应激,而PDE4B无此作用。与非衰竭心脏相比,衰竭人类心脏中PDE4D蛋白表达升高,其中 PDE4D5 变体表达显著增加。通过在NRVMs中过表达PDE4D5,发现PDE4D5可能参与心脏肥大和线粒体损伤的病理过程。进一步研究证实PDE4D5过表达会抑制心肌细胞中PINK1和Parkin的蛋白表达,降低LC3BII/I比值,增加P62表达,而PDE4B过表达对上述指标无明显影响,综合说明线粒体自噬受损与PDE4D诱导的心肌细胞肥大和线粒体损伤有关。
PDE4D,而不是PDE4B,诱导心肌细胞肥大和氧化应激
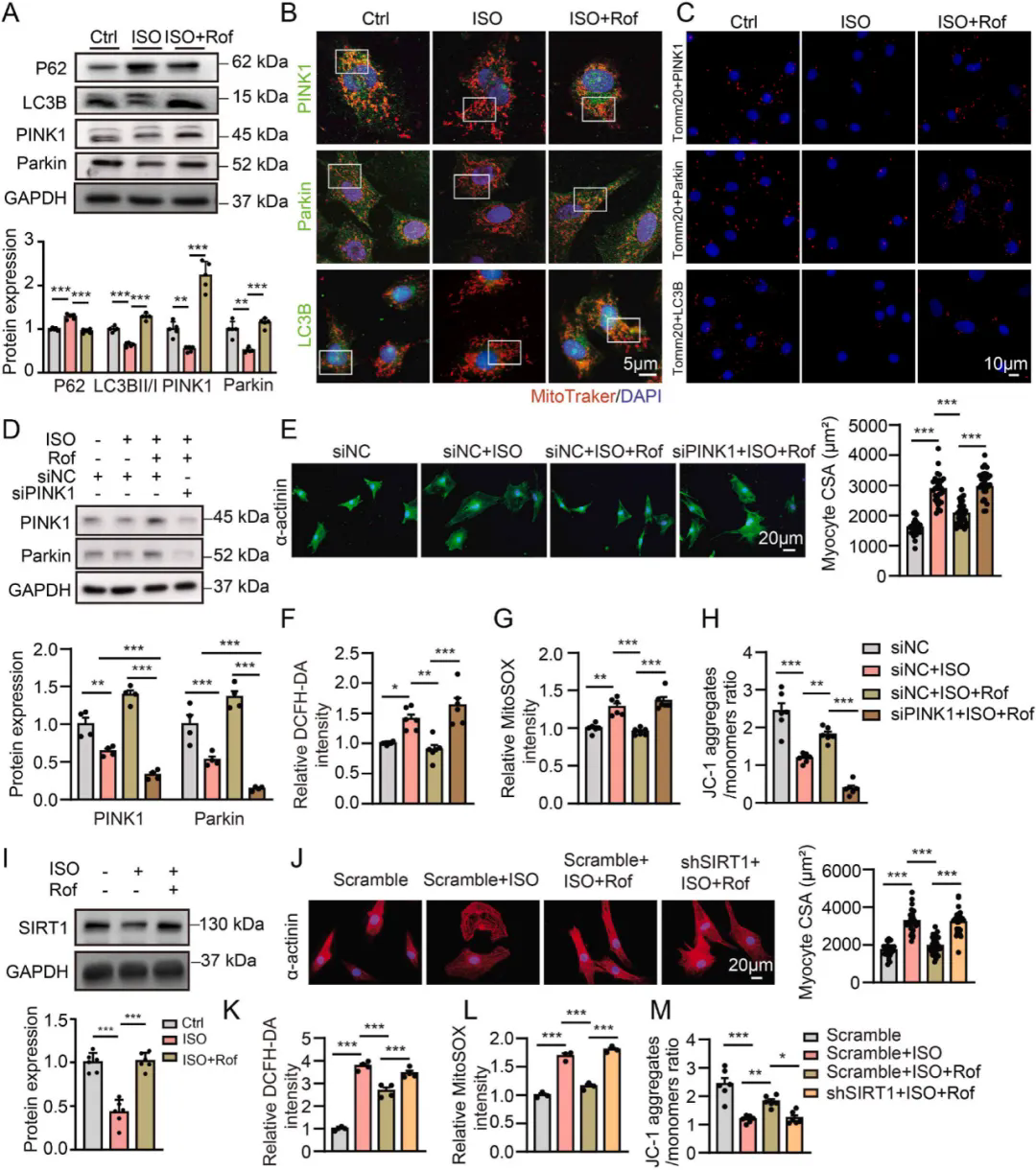
2、PDE4抑制剂通过SIRT1/PINK1/Parkin通路诱导线粒体自噬,保护心肌细胞免受肥大和氧化应激
使用siRNA敲低PINK1表达后,罗氟司特诱导的PINK1和Parkin表达增加被消除,其对ISO处理的NRVMs 细胞大小、氧化应激和线粒体损伤的保护作用也被阻断,且抑制了罗氟司特诱导的Parkin和LC3B与线粒体的共定位。SIRT1可通过改善PINK1/Parkin介导的线粒体自噬保护线粒体。ISO处理显著降低心肌细胞中SIRT1的表达,罗氟司特可减弱这一降低。用SIRT1 shRNA处理后,罗氟司特对ISO诱导的心脏肥大、氧化应激和线粒体损伤的保护作用被消除,其诱导的PINK1、Parkin和LC3B与线粒体的共定位也减弱,表明PDE4抑制剂通过激活SIRT1介导的PINK1/Parkin 信号通路诱导线粒体自噬,发挥心脏保护作用。进一步研究表明PDE4 抑制剂可减轻TAC诱导的心脏肥大和心力衰竭,主要通过促进心脏线粒体自噬实现。
PDE4抑制剂通过SIRT1激活诱导线粒体自噬,防止心肌细胞肥大和线粒体功能障碍
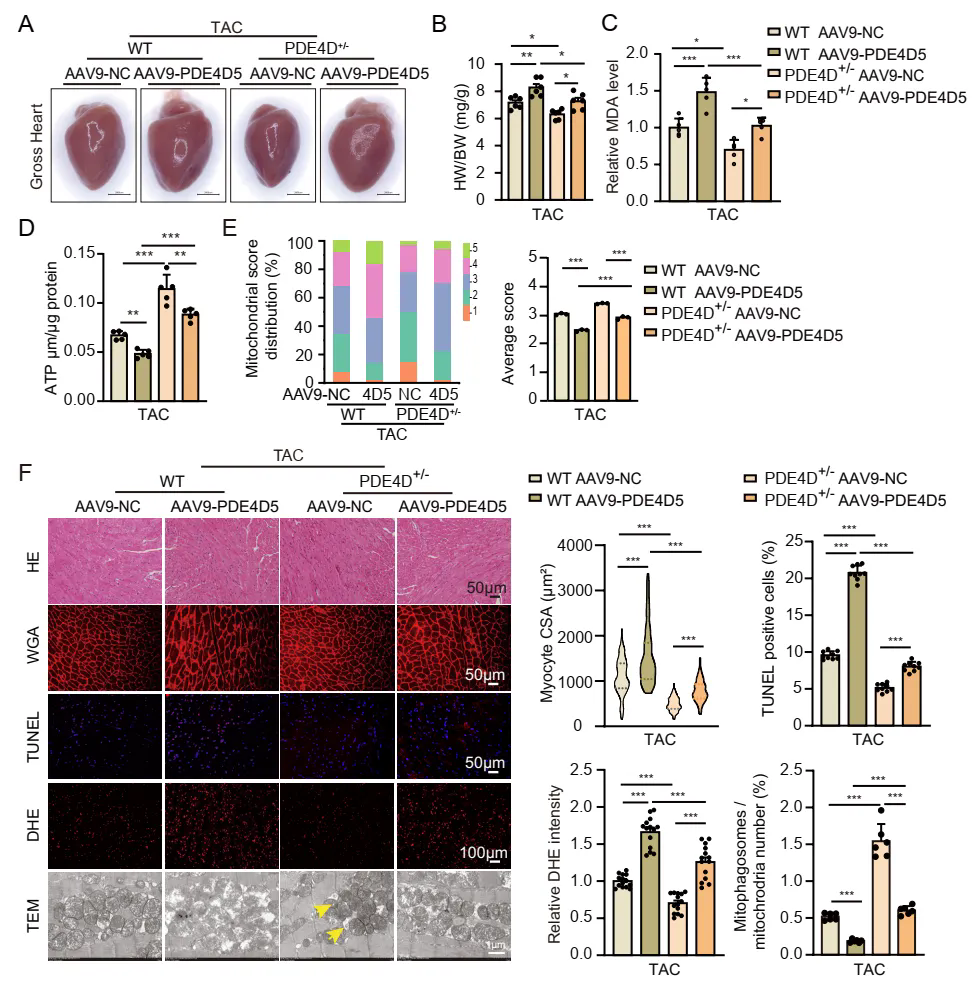
3、PDE4D5的心脏过表达抵消了PDE4D敲除对TAC小鼠的心脏保护作用
选用他莫昔芬诱导的心脏特异性杂合PDE4D敲除(PDE4DhCKO )小鼠,进行TAC手术及他莫昔芬处理,通过评估心脏功能、细胞状态及线粒体自噬情况,证实下调PDE4D对TAC诱导的心脏肥大和心力衰竭有保护作用。为了进一步研究PDE4D5在体内病理性心肌肥大和心力衰竭中的作用,研究团队使用杂合PDE4D基因敲除小鼠(PDE4D+/-),通过静脉注射携带PDE4D5的AAV9载体,使心脏特异性过表达PDE4D5,这一操作加剧了TAC小鼠的心脏肥大和收缩功能障碍,同时恶化了细胞凋亡、ROS水平和MDA含量,还降低了线粒体嵴评分、线粒体自噬体形成和ATP含量。此外,PDE4D5 过表达抵消了PDE4D+/−小鼠因PDE4D基因敲除所产生的保护效果,表明PDE4D5在促进心脏肥大和心力衰竭中起到关键作用,它主要通过抑制线粒体自噬来实现这一过程。
PDE4D5的心脏过表达抵消了PDE4D敲除对TAC小鼠的心脏保护作用
研究结论
本研究阐明持续肾上腺素能激活通过cAMP-PKA信号上调PDE4D表达,抑制 CREB-SIRT1信号介导的线粒体自噬,导致心肌细胞肥大和线粒体损伤。强调抑制PDE4D表达可能是治疗HF的一种新策略。

 当前位置:首页 > 新闻中心 > 新闻资讯
当前位置:首页 > 新闻中心 > 新闻资讯