

 当前位置:首页 > 新闻中心 > 新闻资讯
当前位置:首页 > 新闻中心 > 新闻资讯
索拉非尼 (Sorafenib )是一种口服的多靶点、多激酶抑制剂,可以靶向作用于肿瘤细胞及肿瘤血管上的丝氨酸/苏氨酸激酶及受体酪氨酸激酶等,具有抗血管生成和抗肿瘤细胞增殖的双重作用,已被批准用于晚期肝细胞癌、晚期肾细胞癌和转移性甲状腺癌患者的治疗。对于大多数患者来说,索拉非尼都是一种可以产生良好疗效的药物,但是索拉非尼所诱发的不良反应——高发生率的手足皮肤反应(hand-foot skin reaction,HFSR)仍限制了其临床应用,多项临床试验报告表明,索拉非尼所致的HFSR的发病率在30%到76%不等。
HFSR以掌和足底角化过度(角质层增厚)为特征,通常是由角质形成细胞的表皮稳态异常引起的,包括过度增殖和过度分化,因而角质形成细胞功能障碍在很大程度上被认为是索拉非尼诱导HFSR的主要原因。但是基于这一概念的干预策略后续被研究者证明是无效的,例如,通过预防性去除过度角化区,使用保湿霜来控制足底的过度角化,给患者更换软鞋或者软鞋垫等方式,这些措施对于严重HFSR患者的缓解作用甚微。所以对于重度的HFSR患者,仍需要减少索拉非尼剂量或中断使用,降低所带来的极大的HFSR副反应。但减少或者中断索拉非尼的使用,不仅会降低癌症治疗效果,甚至会导致癌症的恶化。由于目前对药物引发皮肤毒性的潜在机制知之甚少,因此迫切需要基于索拉非尼诱导HFSR的潜在机制开发出有效的干预措施。
2020年,浙江大学和杭州市第一人民医院研究团队在《Cell Research》(IF=20.507)上联合发表了题为“s-HBEGF/SIRT1 circuit-dictated crosstalk between vascular endothelial cells and keratinocytes mediates sorafenib-induced hand-foot skin reaction that can be reversed by nicotinamide”的研究论文。该研究揭示了血管内皮细胞促进角质形成细胞角质化的机制,并为索拉非尼诱发的HFSR提供了潜在的治疗策略。

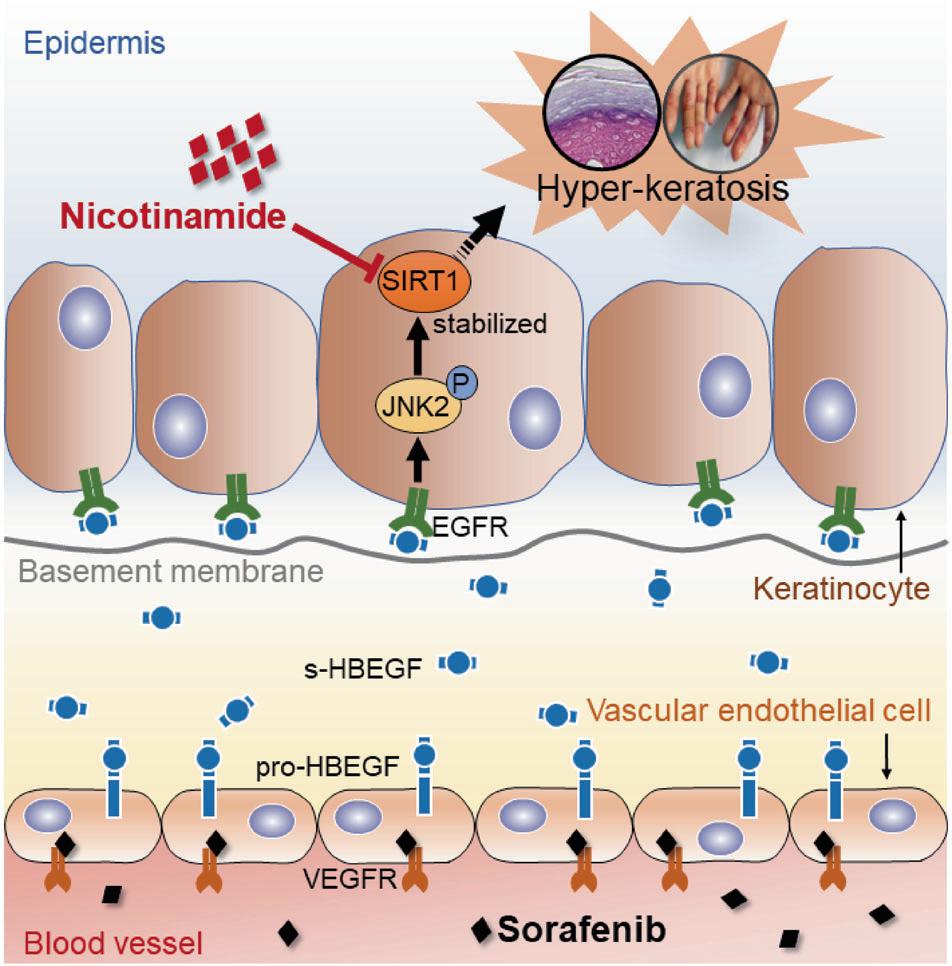
本研究中,研究者证实了血管内皮细胞是索拉非尼诱发HFSR的主要细胞靶点,血管内皮细胞释放的可溶性肝素结合表皮生长因子s-HBEGF激活了角质形成细胞上的表皮生长因子受体(epidergrowth factor receptor,EGFR),促进下游JNK2蛋白的磷酸化,进而稳定了角质形成细胞中必不可少的角化诱导剂SIRT1,并最终引起HFSR的发生。此外,研究人员进一步证明了SIRT1抑制剂烟酰胺可以逆转索拉非尼诱发的HFSR。从体外细胞实验到动物实验,再到初步临床研究,研究人员成功应用SIRT1抑制剂烟酰胺显著改善了10余例服用索拉非尼导致HFSR的患者症状,揭示了烟酰胺可能是一种很有前途的抗索拉非尼诱发HFSR的临床治疗方法。
研究者首先通过人角质形成细胞来测试索拉非尼对角质形成细胞的直接影响。使用人原代角质形成细胞和HaCaT细胞(人永生化角质形成细胞),并以角蛋白5(KRT5)和角蛋白14(KRT14)为细胞增殖标记,以角蛋白1(KRT1)、角蛋白10(KRT10)、兜甲蛋白(LORICRIN)和外皮蛋白(IVL)作为细胞分化标记,测试了索拉非尼对角质形成细胞的直接影响。结果发现,索拉非尼没能直接诱导角质形成细胞增殖,也没有诱导角质形成细胞的分化。这说明,索拉非尼并不直接对角质形成细胞产生影响而引起角化过度。
接着,研究者开始探寻引发HFSR的可能因素。目前已经有报道指出,血管内皮细胞是索拉非尼的细胞靶点之一,在抗血管生成的治疗中,血管内皮生长因子(VEGF)的抗体贝伐单抗显著增加了索拉非尼诱导的HFSR的总发生率。与此同时,作者也通过实验证实了索拉非尼对人脐静脉内皮细胞(HUVECs)的增殖有抑制作用。基于这些研究发现,研究者提出以下假设:血管内皮细胞是否参与了索拉非尼诱导的HFSR。
为了验证这一假设,研究者首先建立了一个模型来研究血管内皮细胞对角质形成细胞的影响,研究者用二甲基亚砜或索拉非尼孵育HUVECs(人脐静脉内皮细胞)24 h,再用该条件培养基(CdMCTRL或CdMSORA)孵育角质形成细胞24 h,研究发现CdMSORA对细胞的增殖没有影响,但角质形成细胞中四种分化标志物((KRT1,KRT10,LORICRIN和IVL)在mRNA表达水平上的表达明显增加,这提示,索拉非尼通过血管内皮细胞诱导了角质形成细胞的分化(图1)。
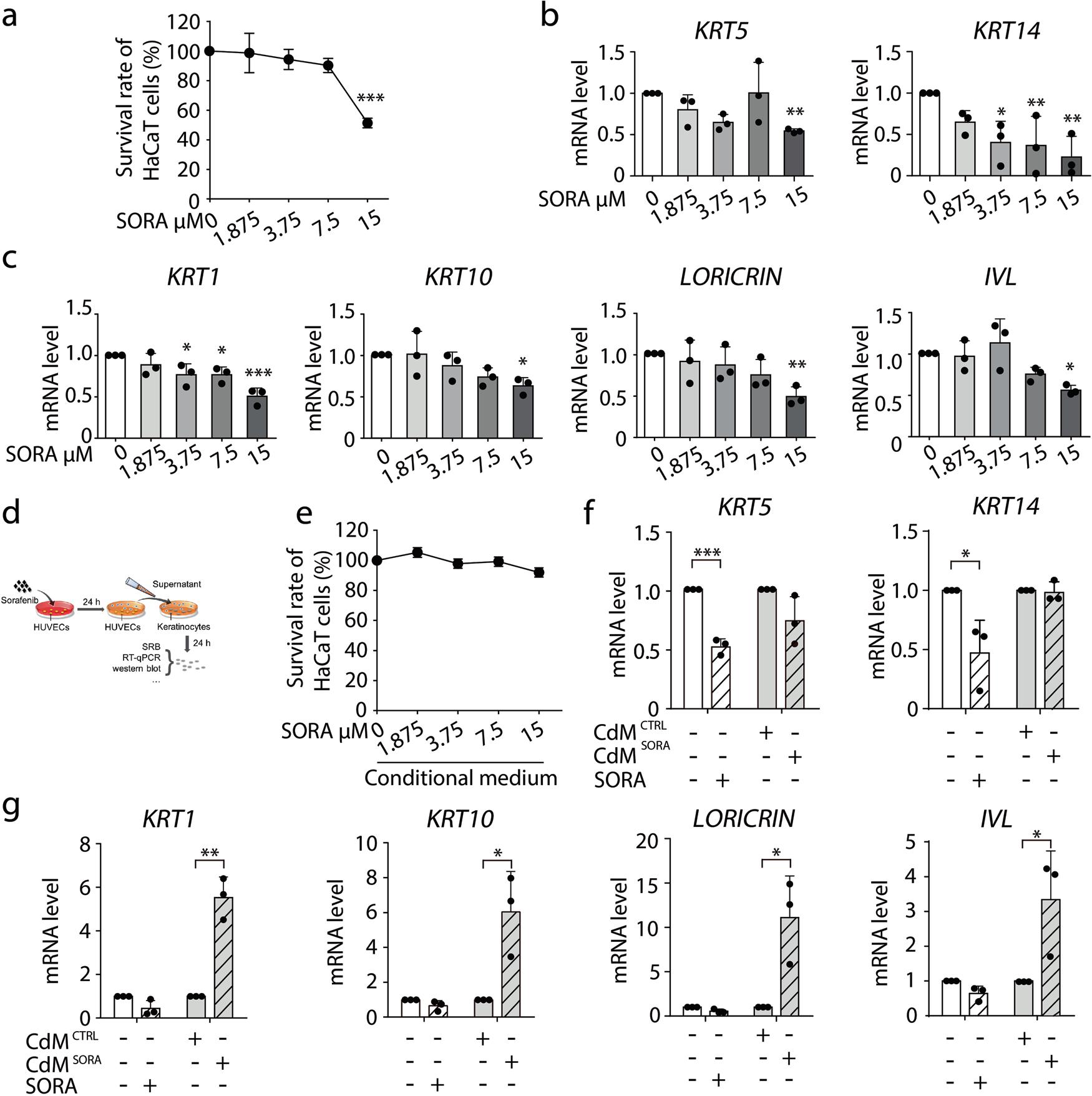
图1. 血管内皮细胞参与索拉非尼诱发的HFSR
接着,研究者又对角质形成细胞分化的驱动因素进行了探究。在用索拉非尼处理的HUVEC培养基中,通过质谱分析检测到了HBEGF,据报道掌跖角化病的发生与s-HBEGF(可溶性的HBEGF)水平升高有关。为了进一步探讨s-HBEGF水平与HFSR严重程度的关系,研究者检测了10例经索拉非尼治疗后确诊的HFSR患者的血清,ELISA结果显示,与健康人相比,HFSR患者的血清中s-HBEGF的表达水平明显增加,并且HFSR越严重,s-HBEGF的水平越高,这说明,s-HBEGF可能在索拉非尼诱发的HFSR中起关键作用。
为了进一步验证s-HBEGF是否确实能够介导索拉非尼诱导的HFSR,研究者进行了一系列体外实验。首先,研究者用s-HBEGF重组蛋白(重组蛋白的浓度与索拉非尼处理的人脐静脉内皮细胞培养液中的浓度成正比)处理人原代角质形成细胞和HaCaT细胞,细胞存活实验显示细胞的增殖几乎没有变化,但是几种细胞分化标记物的mRNA水平明显升高,表明s-HBEGF确实具有促角质化的作用。然后,研究者使用了HBEGF中和抗体阻断s-HBEGF的功能,发现CdMSORA诱导的角化过度被显著逆转,并且HaCaT细胞中四种分化标记物的mRNA水平均显著下降。此外,研究者还构建了HBEGF基因沉默的HUVECs,发现在HBEGF基因敲低的HUVECs中,CdMSORA未能增强索拉非尼诱导的HaCaT细胞角化。
接着,研究者又通过一系列体内试验进行了验证,开发出索拉非尼治疗小鼠模型,实验组小鼠注射HBEGF中和抗体,对照小鼠注射等量的IgG,30天后测量小鼠的角质层厚度。结果发现与给予IgG的索拉非尼组小鼠相比,注射HBEGF中和抗体的索拉非尼治疗组的小鼠,角质层厚度显著降低,这说明HBEGF中和抗体几乎可完全逆转索拉非尼诱发的角质层增厚。接着,研究者又对小鼠模型血清样本中s-HBEGF的浓度进行了检测,与预期一致,索拉非尼治疗组小鼠的s-HBEGF水平显著高于对照组,而抗体治疗显著降低了s-HBEGF的水平。值得注意的是,索拉非尼治疗后表达最厚角质层的小鼠具有最高浓度的s-HBEGF。(图2)。
最后,研究者又探索了在没有索拉非尼存在的情况下,s-HBEGF是否可以诱导HFSR的产生。将静脉注射重组小鼠s-HBEGF蛋白注射入小鼠体内,分析小鼠角质层厚度,发现s-HBEGF特异性组的角质层厚度和分化标志物的表达水平明显高于对照组。这些结果证实了s-HBEGF在体内可以直接诱导角质化的发生。
上述结果表明,s-HBEGF在索拉非尼诱导的HFSR中介导了血管内皮细胞和角质形成细胞之间的相互作用,具体表现为索拉非尼能促进HUVECs释放s-HBEGF,加强角质化,并可进一步导致HFSR的发生。
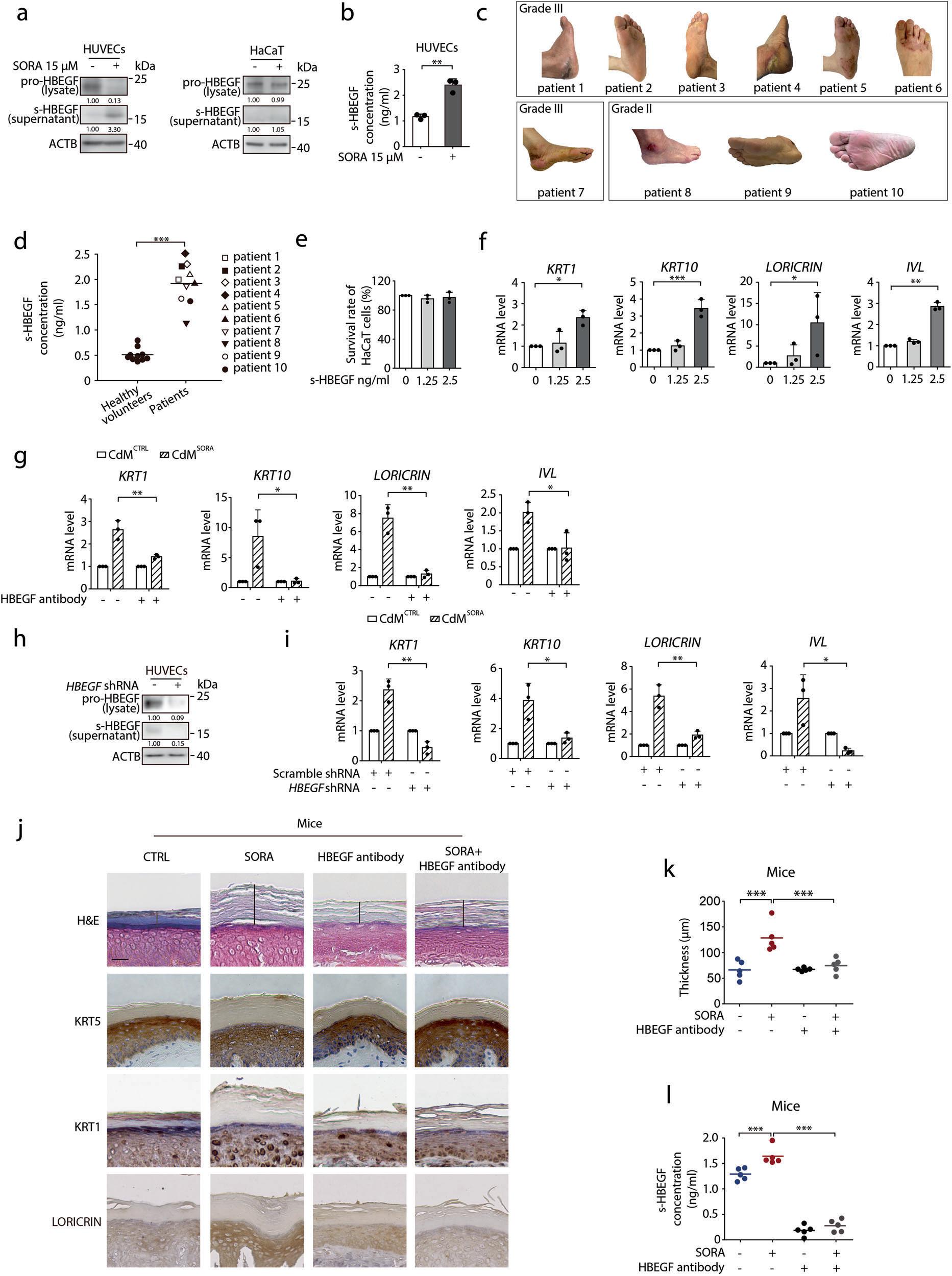
图2. s-HBEGF与索拉非尼诱导的角化过度有关
尽管s-HBEGF被报道参与了许多角化性疾病,但是潜在的调控机制仍然不清楚。有研究表明,s-HBEGF可以通过结合EGFR受体激活下游信号转导,在这一发现的启发下,研究者试图揭开s-HBEGF触发的过度角化病的机制。作者构建了EGFR沉默的HaCaT细胞,检测EGFR对过度角质的影响,发现沉默HaCaT细胞中的EGFR的表达对角质形成细胞的增殖几乎没有影响,但其4种分化标记物的表达水平被逆转,表现为mRNA表达水平的明显下降,表明EGFR作为s-HBEGF受体在索拉非尼介导的角化过度中发挥关键作用。
为了确定s-HBEGF刺激EGFR的效应因子,在s-HBEGF重组蛋白处理的HaCaT细胞中,研究者检测了四个经典的下游通路(AKT,STAT3、MEK和JNK1/2)的表达水平。结果显示,只有磷酸化的JNK(p-JNK1/2)水平升高,而p-JNK1和JNK1水平没有变化。因此研究者又通过JNK2沉默来研究JNK2在s-HBEGF触发的角化过度中的作用,发现在p-JNK1和JNK1水平不变的情况下敲低JNK2时,能显著降低角质形成细胞的分化,而不影响HaCaT细胞的增殖。基于上述研究结果,研究者得出,EGFR-JNK2通路的激活可能是索拉非尼诱导角化过度的重要环节(图3)。

图3. EGFR-JNK2通路参与索拉非尼诱导的角化过度
据报道,JNK2可以磷酸化和稳定SIRT1,而SIRT1与角质形成细胞的分化有关。基于这一点,研究者对SIRT1的表达进行了检测,通过Western blot和免疫组织化学染色观察到,在CdMSORA或s-HBEGF重组蛋白处理的角质形成细胞以及索拉非尼或s-HBEGF处理的鼠爪中,SIRT1的表达水平确有增加。然而相反的是,与索拉非尼治疗组相比,HBEGF抗体和索拉非尼联合治疗组中SIRT1的表达水平降低。为了进一步证实s-HBEGF激活的JNK2可以稳定角质形成细胞中的SIRT1,研究者首先证明了JNK2的敲除抵消了s-HBEGF增强的SIRT1的表达。另外,实验发现s-HBEGF重组蛋白可以延长SIRT1在蛋白合成抑制剂CHX作用下的降解时间,并且可以磷酸化SIRT1第27位丝氨酸,同时抑制其泛素化。敲低JNK2后可以显著逆转s-HBEGF对SIRT1的稳定作用。因此,s-HBEGF激活的JNK2可以稳定角质形成细胞中的SIRT1(图4)。
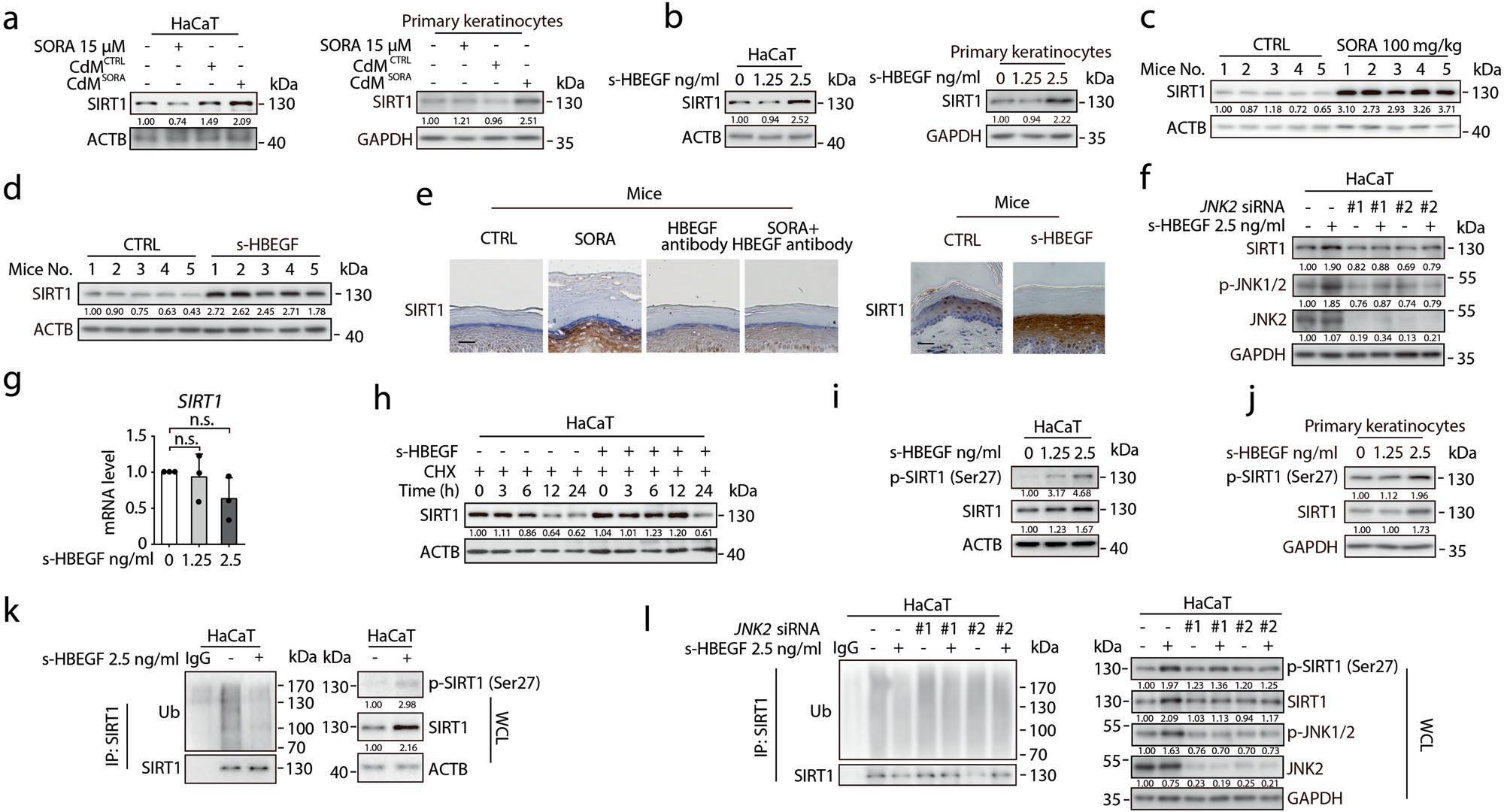
图4. s-HBEGF通过JNK2稳定SIRT1进而诱导角化过度
基于以上研究,作者假设SIRT1被p-JNK2稳定后会导致角化过度,为了证明这一点,作者评估了SIRT1沉默对索拉非尼诱导的过度角化的影响。通过敲低SIRT1,发现CdMSORA或s-HBEGF重组蛋白处理下角质形成细胞的分化有所降低,而不影响细胞的增殖。表明SIRT1的确具有促进索拉非尼诱导的角质形成细胞的分化。
接下来,研究者又进行了体内实验,确定SIRT1在索拉非尼体内诱导HFSR中的意义:将AAV1-shRNA-Sirt1(维真生物提供,病毒滴度为1.98×10E13 vg/mL;每只鼠爪注射2.5×10E11 vg) 通过皮下注射到小鼠爪子中,结果显示Sirt1的敲低逆转了索拉非尼引起的角质层增厚,并且降低了角质形成细胞分化标记物的水平(图5)。
综上所述,JNK2被s- HBEGFf受体EGFR激活后发生磷酸化进而稳定SIRT1引起角化过度。
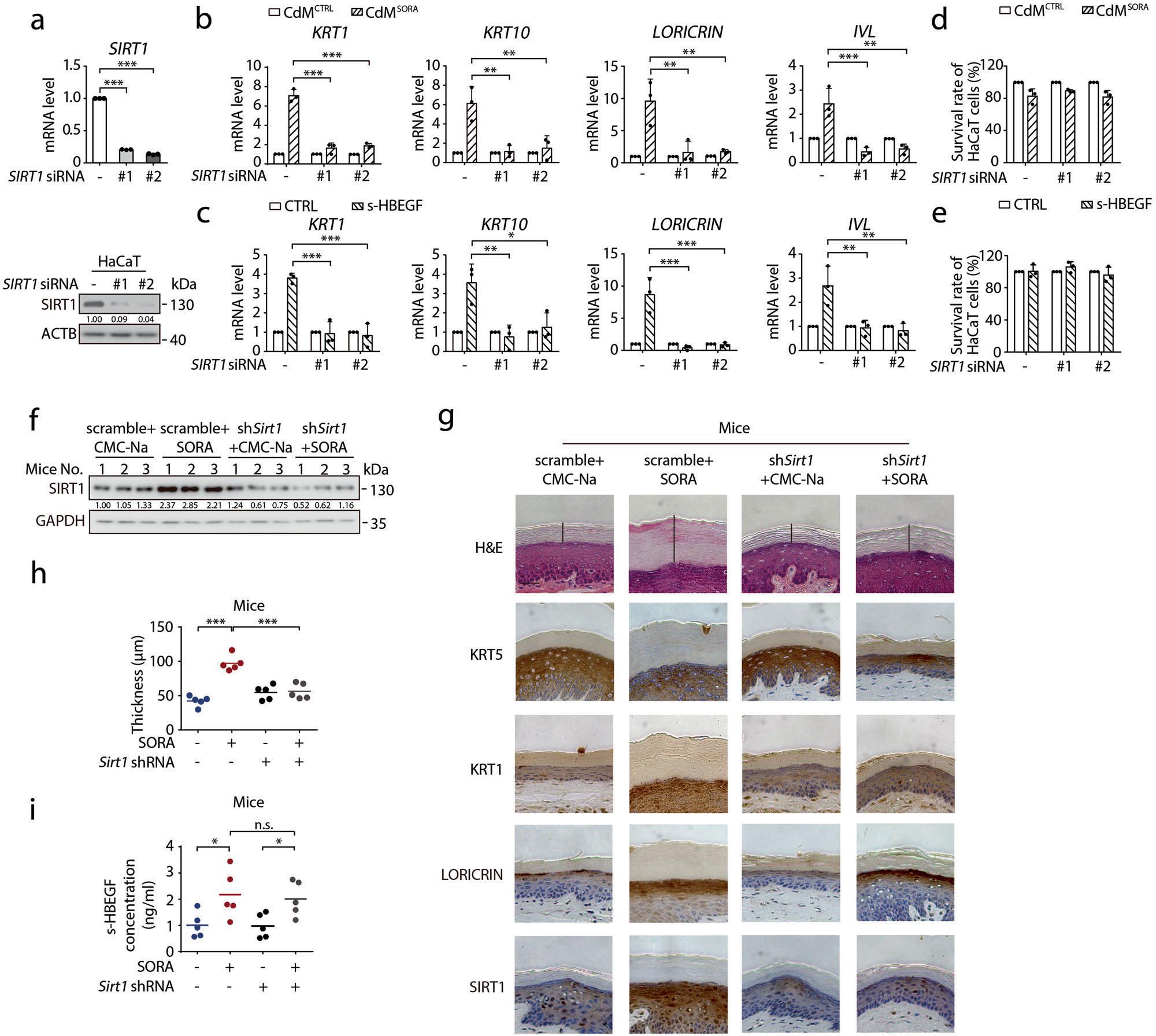
图5. SIRT1参与索拉非尼诱导的角化过度
小结

400-077-2566

service@wzbio.cn







