当前位置:首页 > 新闻中心 > 新闻资讯
当前位置:首页 > 新闻中心 > 新闻资讯
|
病毒产品:AAV Ctrl & AAV-MCL-1 |
注射动物:6月龄雄性转基因APP/PS1 (C57BL/6)小鼠 |
|
病毒用量:1.5 μL (0.15 μL/min) |
注射方式:立体定位注射 |
|
注射部位:海马区 |
检测时间:30天后 |
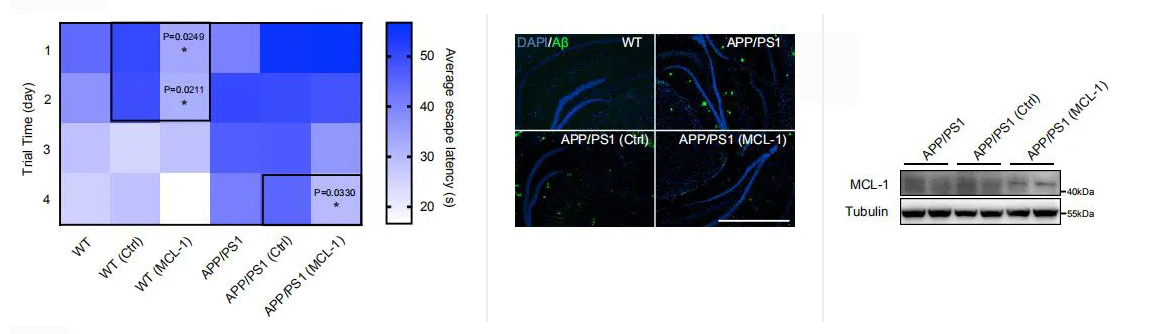
作者以阿尔兹海默症转基因小鼠模型APP/PS1为研究对象,将AAV-MCL-1过表达载体注射至小鼠的海马区,结果显示,过表达MCL-1基因能显著改善APP/PS1小鼠的认知功能,而且海马区细胞外的Aβ斑块出现减少。此外,MCL-1的过表达也提高了野生型小鼠的学习和记忆能力,这表明MCL-1能在神经元中发挥非常重要的作用。
阿尔茨海默病(Alzheimer’s disease ,AD)是一种由神经元死亡而造成的神经变性疾病——以进行性记忆力减退和获得性知识丧失,直至日常生活活动能力完全丧失为特征,给社会和家庭带来沉重负担,成为严重的社会和医疗卫生问题。阿尔茨海默病是继心血管病、脑血管病和肿瘤之后,威胁老年人健康的重要疾病。目前,全球约有5000万人罹患阿尔兹海默症。预计到2050年,这个数字将增加至1.52 亿。当前,全球每年用于治疗、护理阿尔兹海默症病人的费用已经达到1万亿美元,而这一数字将在2030年达到目前的两倍。
阿尔茨海默病的病因复杂,目前主流观点认为是由β淀粉样蛋白(Aβ)和微管相关蛋白Tau沉积造成神经元大量死亡引发的,1998年以来,有100余种治疗此病的药物进行临床试验,但仅有6种针对此病症的药物获得FDA的批准上市,而且近年来世界各大制药公司针对Aβ或Tau蛋白开发的药物均遭到了不同程度的失败,这给人类对于AD的攻克埋上了一层巨大的阴影。
线粒体功能障碍是AD的一个基本病理特征,在散发性和家族性AD病例以及AD动物模型中,均被发现有受损的神经元线粒体的积累。功能受损的线粒体会触发能量应激,从而促进Aβ寡聚化和Tau过度磷酸化。线粒体合成调节因子PGC-1α的表达变化以及线粒体功能失调导致的钙稳态失衡都已被证实与AD的发生有关。新的研究表明,AD患者的脑细胞中线粒体自噬受到损害,能造成大量损伤性线粒体积累,导致突触功能障碍和认知功能的下降。因此,保障AD患者神经细胞中线粒体自噬的正常进行至关重要,而找到一种诱导线粒体自噬的药物靶点则是重中之重。
近期,浙江大学基础医学院夏宏光教授团队在《Nature communications 》(IF=12.121)上在线发表了一篇关于阿尔茨海默病的最新研究成果—Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model。该研究首次揭示了抗凋亡蛋白MCL-1作为线粒体自噬受体蛋白介导线粒体自噬的新机制,MCL-1的特异性抑制剂UMI-77可以在AD模型小鼠中显著缓解阿尔茨海默病的病理特征,改善小鼠认知;本研究提出靶向MCL-1蛋白诱导线粒体自噬是一种有巨大前景的治疗阿尔茨海默症的策略。
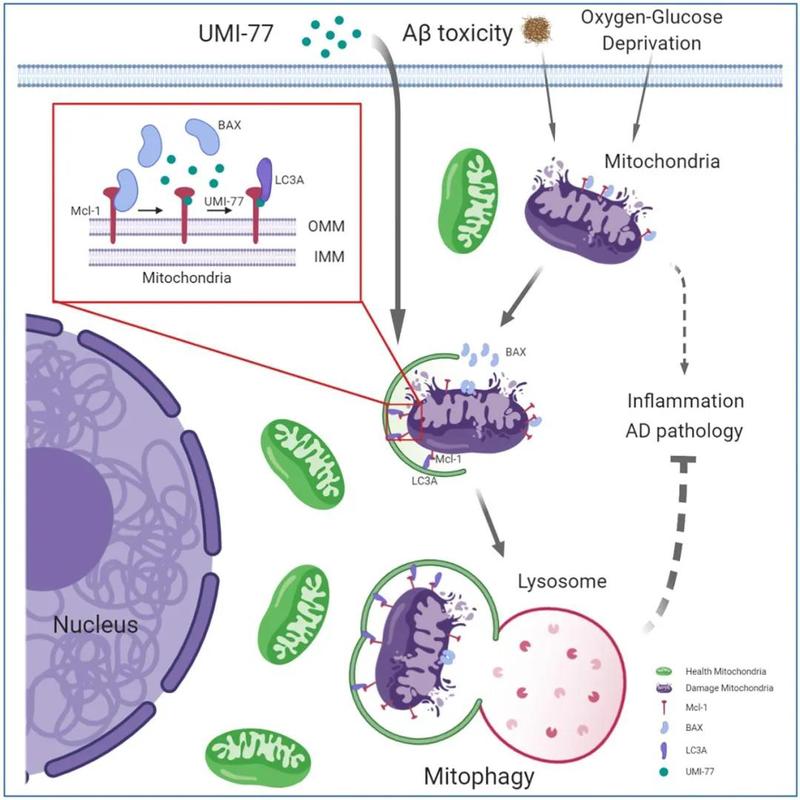
UMI-77作用机制示意图
首先,研究人员通过利用荧光蛋白Keima构建敏感的线粒体自噬定量检测方法,对稳定表达mt-Keima(Keima蛋白具有在酸性和中性pH中荧光信号不同的特性,定位于线粒体中的Keima(mt-Keima)可显示通过自噬途径进入溶酶体中的线粒体,直观地反映线粒体自噬程度)的HEK293T细胞系和一个包含2024个FDA批准的药物或候选药物库进行小分子化合物高通量筛选,找到了一种可以安全有效诱导线粒体自噬的小分子化合物——UMI-77,一种Bcl-2家族抑制剂。
研究表明,UMI-77是抗凋亡蛋白MCL-1的特异性抑制剂,能阻断MCL-1和Bax/Bak之间的相互作用,从而允许Bax/Bak诱导细胞凋亡。研究者通过测定线粒体的去极化及自噬水平,发现与CCCP(氧化磷酸化抑制剂,影响线粒体的蛋白合成)相比,UMI-77在亚致死剂量下不会诱导HEK293T和HeLa细胞的线粒体损伤;在HEK293T-mt-Keima中,UMI-77+CCCP联合使用明显增强了线粒体的自噬水平,表明UMI-77不会造成线粒体损伤,且能够促进线粒体自噬,且能增强CCCP诱导的线粒体损伤引发的线粒体自噬。HEK293T-mt-Keima细胞的活细胞成像实验、透射电子显微镜(TEM)观察及western bloting 实验也证实了UMI-77的促线粒体自噬作用。
为排除凋亡诱导剂对线粒体自噬可能的影响,研究者又做了评估实验。结果发现,药物库中大多数诱导细胞凋亡的药物及pan-caspase抑制剂Z-VAD-fmk(可阻断细胞凋亡)不会影响UMI-77的促线粒体自噬作用,这表明UMI-77诱导的线粒体自噬,不依赖于凋亡诱导,且UMI-77在亚致死剂量下不会诱导细胞凋亡。与此同时,UMI-77处理后,巨自噬标记物p62和其他细胞器标记物的表达水平并没有下降,这表明UMI-77可特异性诱导线粒体自噬,不影响非选择性自噬。
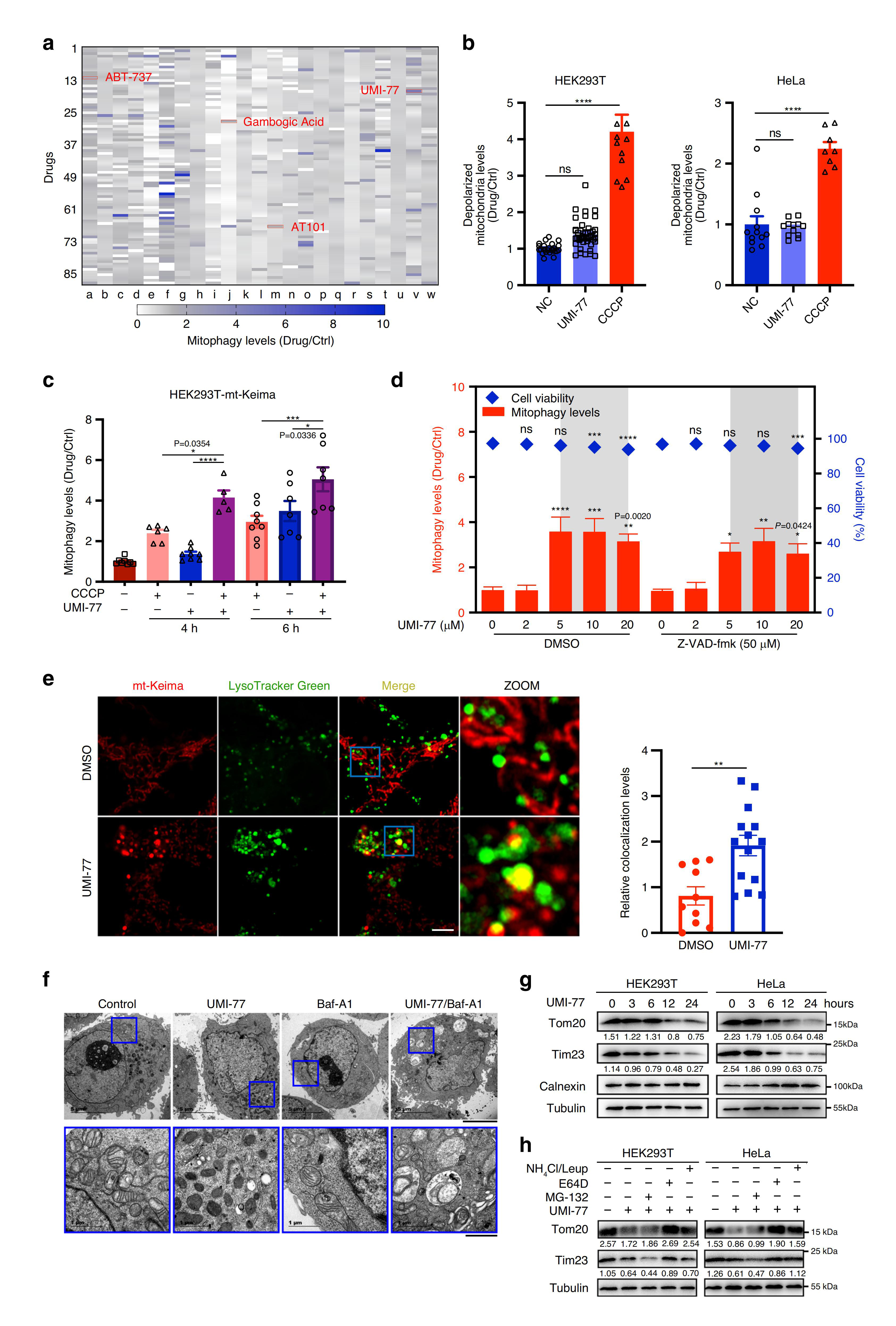
图1. UMI-77可特异性诱导线粒体自噬
既然UMI-77能诱导线粒体自噬,MCL-1又是UMI-77的靶点,那么MCL-1是否参与了UMI-77诱导的线粒体自噬的过程呢?研究者对此进行了探索,发现在HEK293T和HeLa细胞中敲除MCL-1基因后减缓了UMI-77诱导的线粒体蛋白Tom20和Tim23的降解,抑制了UMI-77诱导的线粒体自噬水平,这说明MCL-1在UMI-77诱导的线粒体自噬激活中是必须的。
此前已有研究证明,细胞自噬受体如FUNDC1和FKBP8的过表达能促进细胞自噬,那么MCL-1在线粒体自噬中是否也是扮演自噬受体的角色?为了探究其在自噬中所起的作用,研究者建立强力霉素Dox诱导的MCL-1过表达的HEK293T稳定细胞系(HEK293T- MF2),值得注意的是,HEK293T- MF2中线粒体标志物Tim23的表达水平下降,线粒体变小、片段化,而且线粒体和溶酶体出现了共定位。透射电镜分析也证实了MCL-1的过表达促进了线粒体自噬。综上结果表明,MCL-1在线粒体自噬中发挥关键作用。
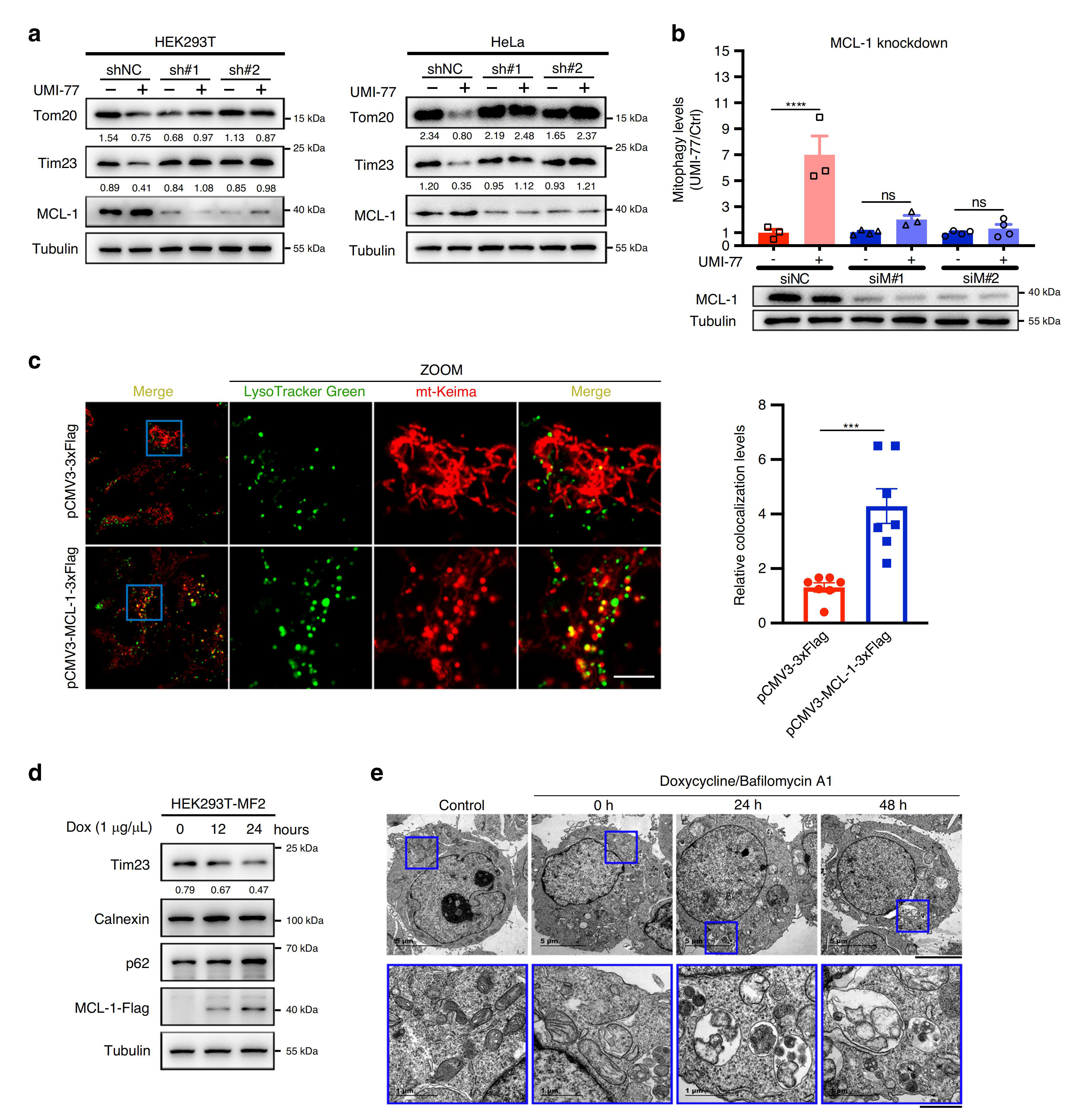
图2. MCL-1促进线粒体自噬
研究者对MCL-1在线粒体自噬中是否扮演自噬受体的角色继续展开研究,并推测UMI-77诱导Bax/Bak中MCL-1释放后能MCL-1能与LC3发生相互作用,从而促进线粒体自噬。研究发现MCL-1在其C端有三个“LC3-相互作用区(LIR)”基序,其中前两个基序LIR261-264和LIR318-321位于胞质区,并且LIR261-264具有很强的保守性,这提示它是有一定功能的。
研究人员首先验证了UMI-77是否能诱导MCL-1和LC3发生相互作用,通过免疫共沉淀试验发现,经UMI-77处理后,LC3A与MCL-1的相互作用增强,而Bax与MCL-1的相互作用随之减弱;在UMI-77的作用下,MCL-1还与其他Atg8家族蛋白相互作用。此外,pull-down实验也显示MCL-1能直接与LC3A结合。随后,研究者又生成了一系列MCL-1 LIR261-264和LIR318 - 321基序的突变体,结果显示MCL-1 LIR261-264基序突变后减弱了MCL-1与LC3A的相互作用。
接下来,研究者对UMI-77是否能增强内源性MCL-1和LC3A的相互作用展开了相关研究:结果与我们预期一致,发现用UMI-77处理后,Duolink®PLA结果显示,在线粒体上能观察到内源性MCL-1和LC3A相互作用的增强,而未用UMI-77处理时这种相互作用也能观察到,反映了基础内源自噬的水平。这个结果进一步表明MCL-1是一个重要的线粒体自噬受体。
最后,作者又验证了MCL-1和LC3A的相互作用是否能在UMI-77介导的线粒体自噬激活中起作用。结果显示,敲除MCL-1后,HEK293T-mt-Keima和SH-SY5Y细胞中UMI-77介导的线粒体自噬水平降低,而补充MCL-1/LIR318-321突变体MCL-1的表达可逆转此现象。
综上所述,MCL-1是一个线粒体自噬受体,它通过其LIR261-264基序与LC3A直接作用,并且这种相互作用能被UMI-77增强进而导致线粒体自噬水平的增强。此外,MCL-1和LC3A之间的相互作用是UMI-77介导线粒体自噬激活的关键。
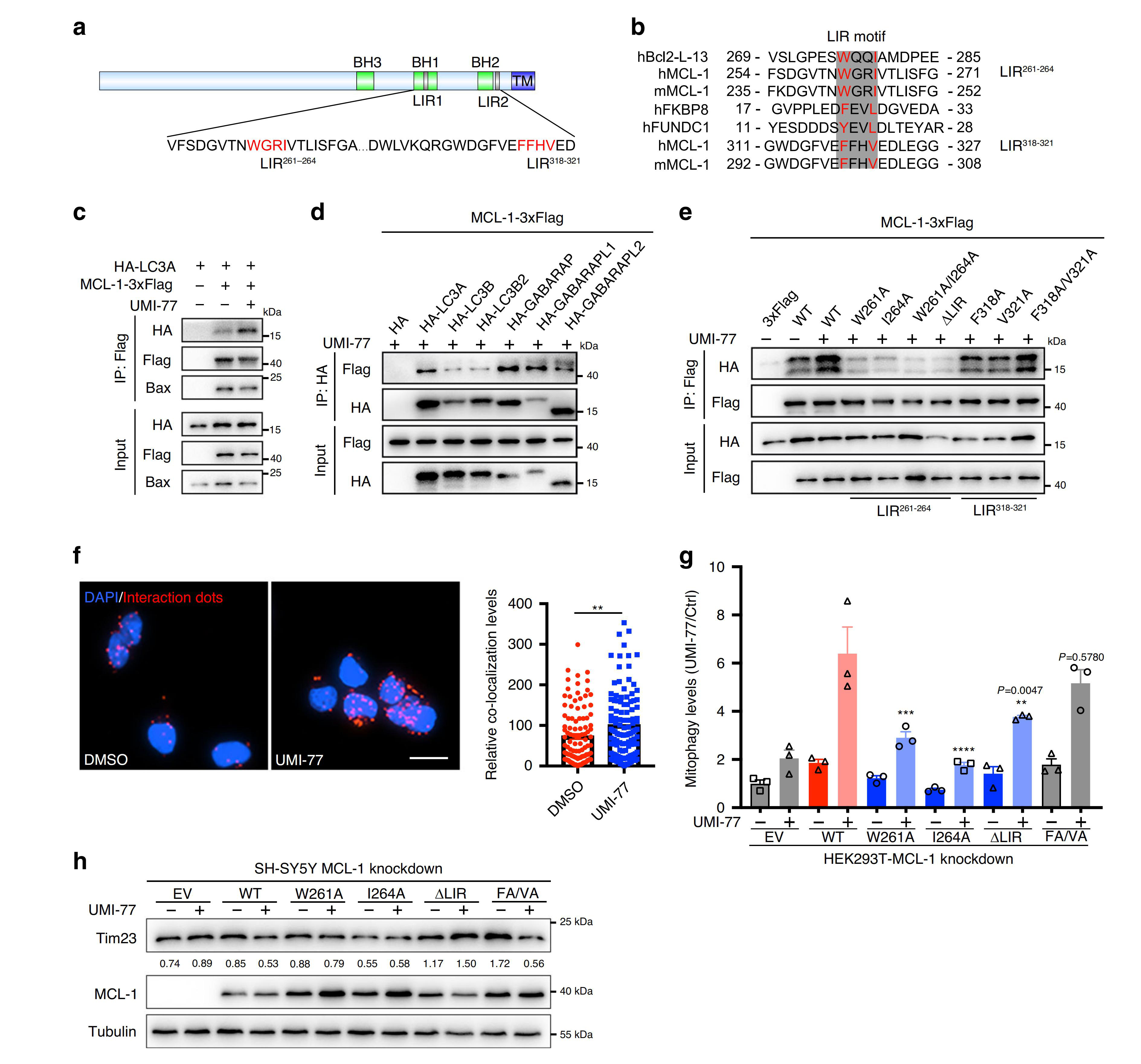
图3. MIC-1和LC3A的相互作用是UMI -77诱导线粒体自噬所必需的
随后,作者研究了其他线粒体自噬受体蛋白是否在UMI-77诱导的线粒体自噬或MCL-1-LC3A相互作用的诱导中发挥作用。作者以野生型HeLa细胞和四基因敲除的突变体HeLa细胞(敲除线粒体自噬受体NDP52、p62、NBR1和TAX1BP1)为研究对象进行研究,发现UMI-77能显著增强MCL-1和LC3A之间的相互作用,线粒体标记蛋白Cox II和Tim23的表达水平也均呈现时间依赖性的降低,此外,线粒体自噬受体(FUNDC1, BNIP3和NIX)也被证明不参与UMI-77诱导的线粒体自噬,表明UMI-77诱导的线粒体自噬与这些线粒体自噬受体蛋白无关。
鉴于UMI-77阻断了MCL-1与Bax的相互作用,研究者随后分析了Bax在UMI-77诱导的线粒体自噬中的作用,令人惊讶的是,敲低Bax后UMI-77诱导的线粒体自噬水平增强,说明Bax不参与UMI-77诱导的线粒体自噬。另外两种MCL-1相互作用蛋白Beclin1和Parkin也先后被排除参与UMI-77介导线粒体自噬激活的可能性。相反,在HEK293T-mt-Keima细胞或MEF细胞中敲除ATG5后抑制了Tom20和Tim23降解,说明ATG5是UMI-77诱导线粒体自噬必需的。
综上结果表明,UMI-77诱导的线粒体自噬是通过ATG5自噬通路介导的,不依赖于线粒体自噬受体蛋白NBR1、TAX1BP1、p62、NDP52、FUNDC1、BNIP3、NIX以及MCL-1相互作用蛋白Bax、Beclin1和Parkin。
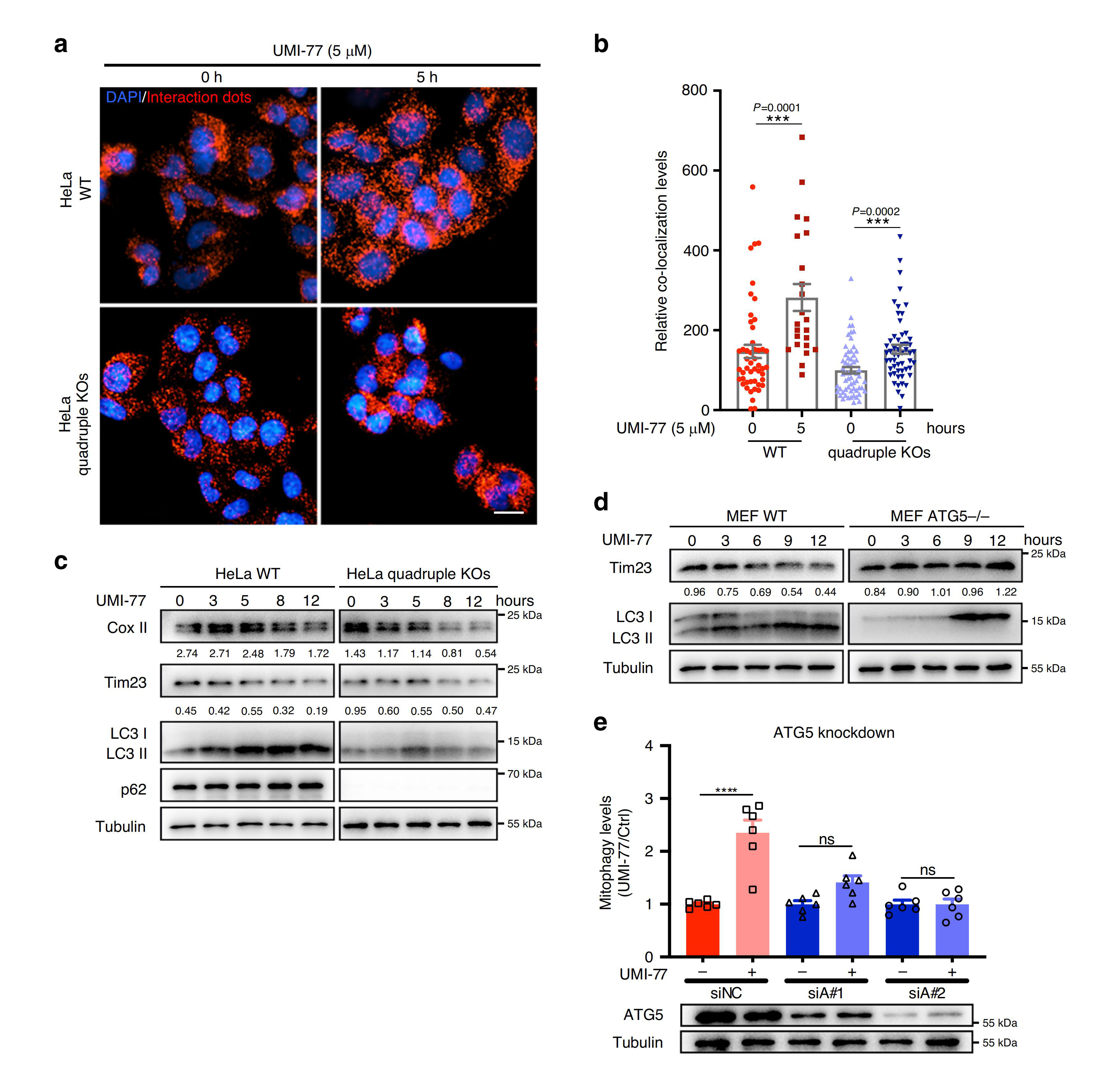
图4. UMI-77以ATG5依赖的方式诱导线粒体自噬
接着,研究者又研究了MCL-1作为线粒体自噬受体的生理功能。研究表明,OGD(氧-糖剥夺)或OGD/再灌注会损伤线粒体,并通过线粒体自噬诱导清除受损线粒体。mt-Keima实验结果显示,敲除MCL-1后OGD诱导的线粒体自噬水平显著降低,线粒体标记蛋白Cox II和Tim23的特异性降解也被抑制,说明MCL-1对OGD诱导的线粒体自噬是必需的。
已有研究证明MCL-1可调节线粒体的碎片化,因此研究人员试图了解MCL-1在OGD诱导的线粒体自噬中的作用。免疫荧光显微镜观察到OGD处理后HEK293T细胞线粒体碎裂,而敲除MCL-1改变了这些形态学变化,表明MCL-1在OGD诱导的线粒体碎片化中起着关键作用,并且MCL-1 LIR261-264基序仅参与线粒体自噬,而不参与MCL-1的线粒体碎片化调节作用。同样地,OGD也增强了MCL-1与LC3A的相互作用,降低了其与Bax的相互作用,再次证明MCL-1是一种线粒体自噬激活的受体。
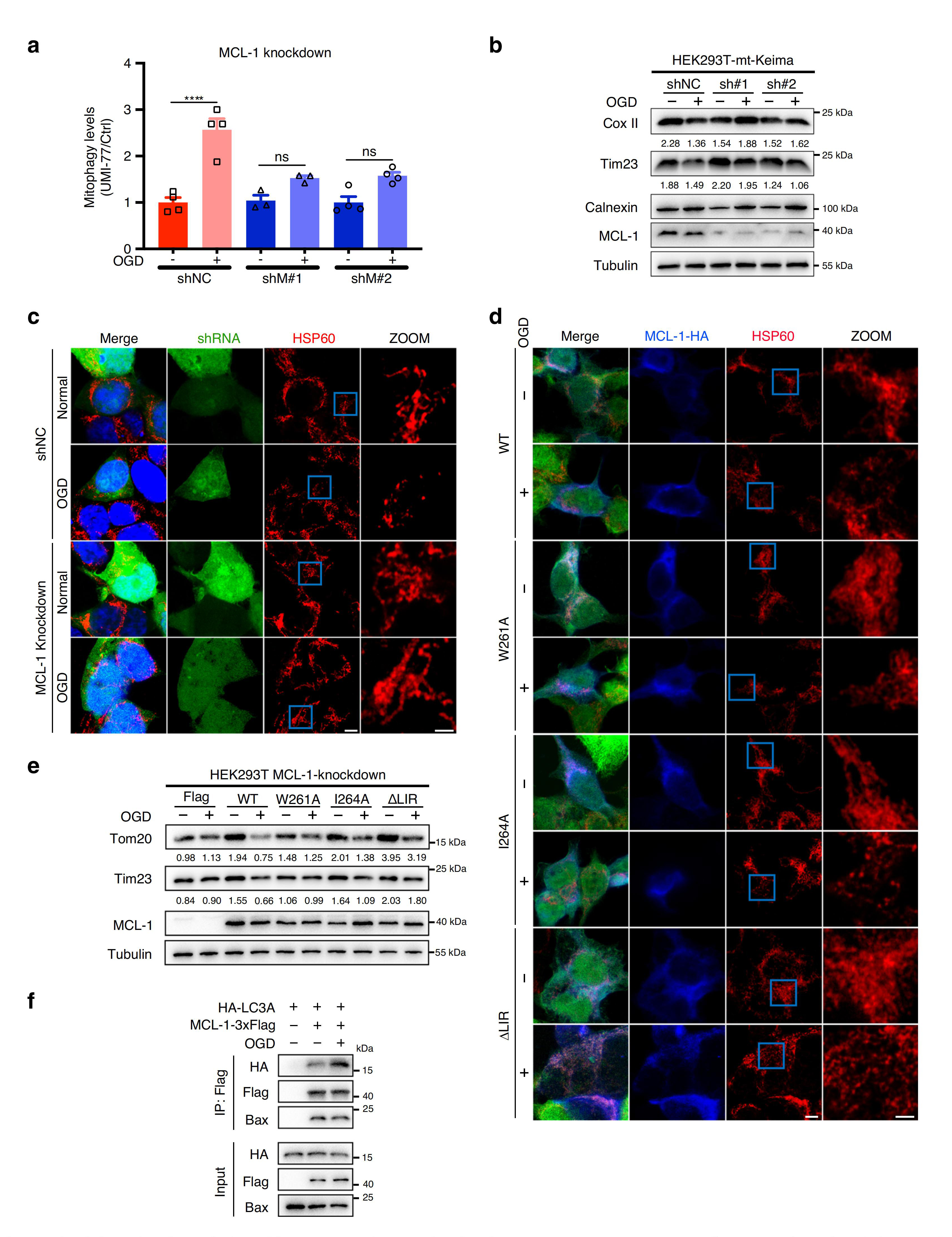
图5. MCL-1是OGD诱导的线粒体自噬所必需的
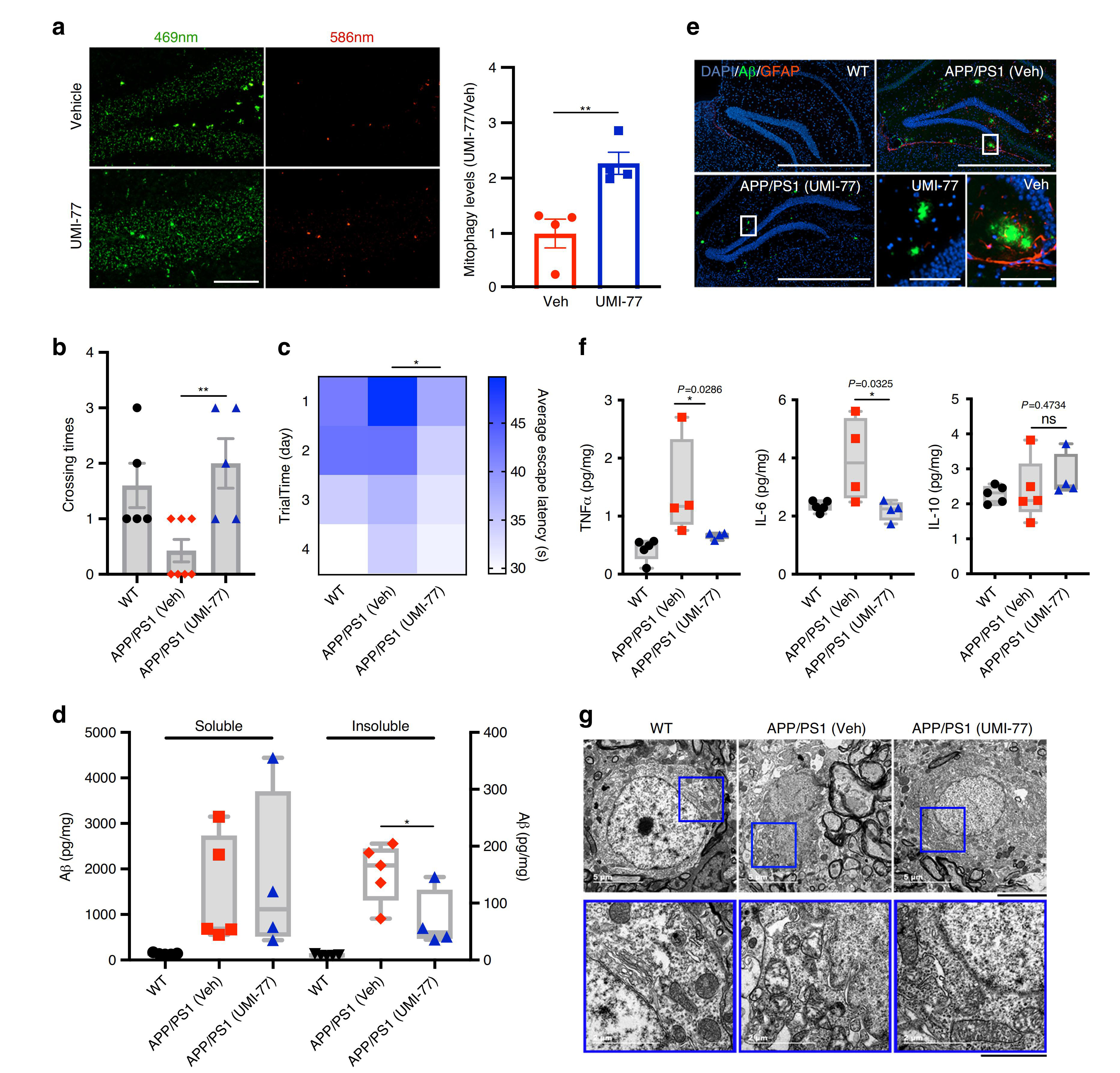
接着,研究人员探究了UMI-77诱导的线粒体自噬对AD模型APP/PS1小鼠疾病病理和行为表型的影响。对4月龄的小鼠腹腔注射10 mg/kg剂量的UMI-77,共持续4个月。通过Morris水迷宫测试发现UMI-77治疗明显改善了APP/PS1小鼠的学习和记忆能力,且能有效降低小鼠脑内不溶性Aβ1-42的水平。同样,免疫荧光结果也显示UMI-77治疗后海马区细胞外Aβ斑块明显缩小,星形胶质细胞的激活也受到抑制。此外,UMI-77也降低了APP/PS1小鼠的神经炎症水平,表现为炎症细胞因子(TNFα和IL-6)水平显著降低。重要的是,UMI-77显著恢复了神经元的线粒体形态,这一研究结果与在APP/PS1小鼠中观察到的通过UMI-77可以诱导线粒体自噬从而清楚受损线粒体相一致。这些结果均表明UMI-77是一种治疗AD的有效药物。
图6. UMI-77是一种治疗AD的有效药物
最后,研究人员评估了MCL-1介导的线粒体自噬对APP/PS1小鼠行为表型的影响。将AAV-MCL-1过表达载体注射小鼠的海马区,MCL-1的过表达显著改善了APP/PS1小鼠的认知功能,并减少了海马区细胞外的Aβ斑块。令人惊讶的是,MCL-1的过表达也提高了野生型小鼠的学习和记忆能力,表明MCL-1在神经元中有非常重要的作用。
综上所述,MCL-1是一种新的线粒体自噬受体蛋白,是治疗阿尔茨海默症的新药物靶点;UMI-77可以通过释放游离的MCL-1蛋白诱导线粒体自噬,显著恢复APP/PS1 AD小鼠模型的认知功能缺损,减轻炎症反应和Aβ斑块引起的病理效应,促进受损线粒体的清除。总而言之,诱导线粒体自噬是治疗阿尔茨海默症的有效策略。

400-077-2566

service@wzbio.cn