

 当前位置:首页 > 新闻中心 > 新闻资讯
当前位置:首页 > 新闻中心 > 新闻资讯
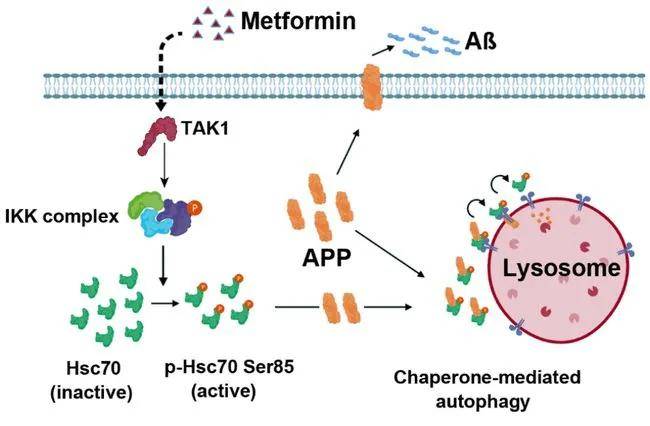
近期,浙大夏宏光教授课题组在《Protein&Cell》(IF=14.971)上发表了题为“Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model”的研究成果,文章首次描述了二甲双胍诱导CMA(分子伴侣介导的自噬)的机制,即通过激活TAK1-IKKα/β信号通路,使热休克蛋白Hsc70 Ser85发生磷酸化并被激活从而调控CMA进程。本研究发现APP是CMA的底物,能以IKKα/β依赖的方式与Hsc70结合,抑制CMA介导的APP降解能增强细胞毒性。此外,在APP/PS1 AD小鼠模型中,Hsc70过表达或二甲双胍激活CMA可以有效降低脑区Aβ斑块积累水平,并逆转AD的分子和行为表型。该研究确定了二甲双胍可作为CMA的新型激活剂,并可作为治疗AD的有效药物,同时也为治疗CMA相关疾病提出了一种有利证据。
病毒产品:AAV8-CAG-mCherry;
AAV8-CAG-Hsc70WT-Flag-P2A-mCherry;
AAV8-CAG-Hsc70(S85A)-Flag-P2A-mCherry;
种属:16周龄APP/PS1小鼠
注射方式:立体定位注射小鼠海马区
注射量:1.5μL ,0.3μL/min
检测时间:注射后12周
阿尔茨海默病(Alzheimer 's disease, AD)是一种常见的神经退行性疾病,临床表现为进行性认知功能障碍和记忆力减退等,其病理特征包括Aβ蛋白沉积和Tau蛋白过度磷酸化。已知Aβ蛋白来源于淀粉样前体蛋白(APP),CMA激活能够降低Tau蛋白水平,因此利用药物促进APP的清除,并寻找安全有效的CMA激活药物,可能是治疗AD的潜在策略。然而CMA在调节Aβ蛋白水平中的作用尚未被证实,此外,目前也缺乏一种能安全有效激活CMA的药物,阻碍了通过CMA激活策略治疗相关疾病可行性的研究。二甲双胍是一种广泛用于治疗2型糖尿病的口服药物,安全性高,研究表明二甲双胍不仅对一些癌症具有有益作用,还可以抑制慢性炎症、抗衰老,并可能保护认知功能。但是二甲双胍是否可用于阿尔茨海默病的有效治疗,目前尚不明确。
首先,本研究通过药物筛选确认二甲双胍是一种新型的CMA激活剂,可以激活IKKα/β激酶,进而磷酸化并激活Hsc70。此外,作者通过体内实验发现,二甲双胍能降低APP/PS1的AD小鼠模型中Aβ水平,进而改善小鼠的认知障碍。最后,作者利用AAV载体介导Hsc70在APP/PS1的AD小鼠海马区的过表达,发现Hsc70过表达能够显著减轻AD的病理过程。
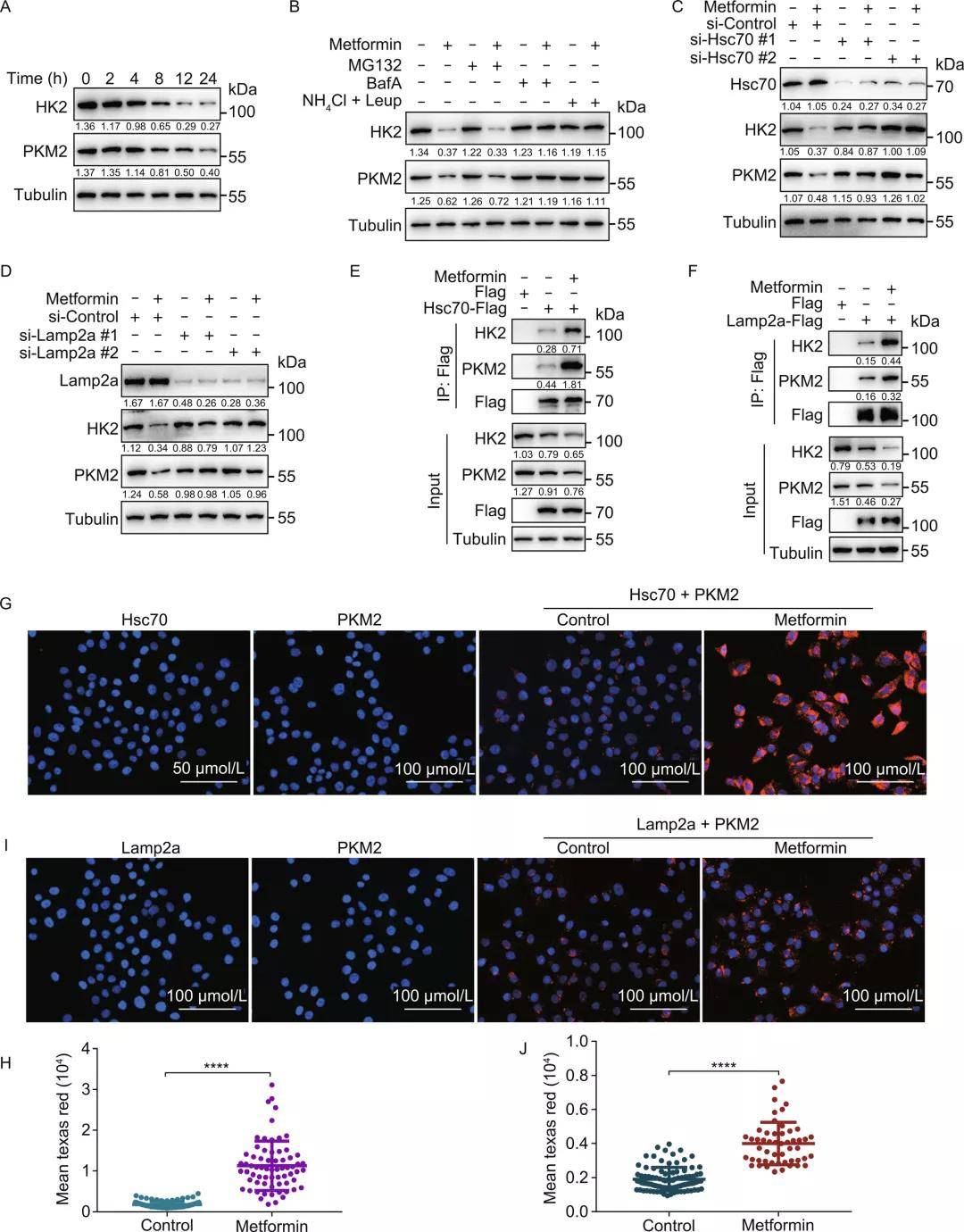
首先,研究者利用293THK细胞(监测CMA活性的功能性报告系统)和高通量流式细胞术筛选了能有效激活CMA的药物。在2197种FDA批准的药物或候选药物中,筛选出了广泛用于治疗2型糖尿病的药物二甲双胍,二甲双胍在Lamp2a(溶酶体受体,介导CMA底物进入溶酶体)正常表达情况下能诱导HK2-GFP水平下降,而在Lamp2a敲低时则不能。
用二甲双胍处理293THK细胞后,HK2-GFP蛋白水平下降,并呈现一定的剂量依赖性,而下调Hsc70表达则逆转了二甲双胍对HK2-GFP的影响。此外,二甲双胍还能诱导两种内源性CMA底物HK2和PKM2的降解,并可被溶酶体抑制剂(如E-64D,BafA等)而非蛋白酶抑制剂所抑制,证明此降解过程依赖于溶酶体。而Hsc70和Lamp2a的敲低阻断了二甲双胍诱导的内源性HK2和PKM2的降解,pull-down和PLA技术均证实二甲双胍诱导内源性HK2和PKM2与Hsc70 和 Lamp2a相互作用,此外还排除了宏自噬在此过程中的作用,证实这些蛋白的降解依赖于CMA。
上述研究结果表明,二甲双胍能够以溶酶体、Hsc70和Lamp2a依赖的方式激活CMA,触发其底物的降解,并促进它们与Hsc70和LAMP2A的相互作用。
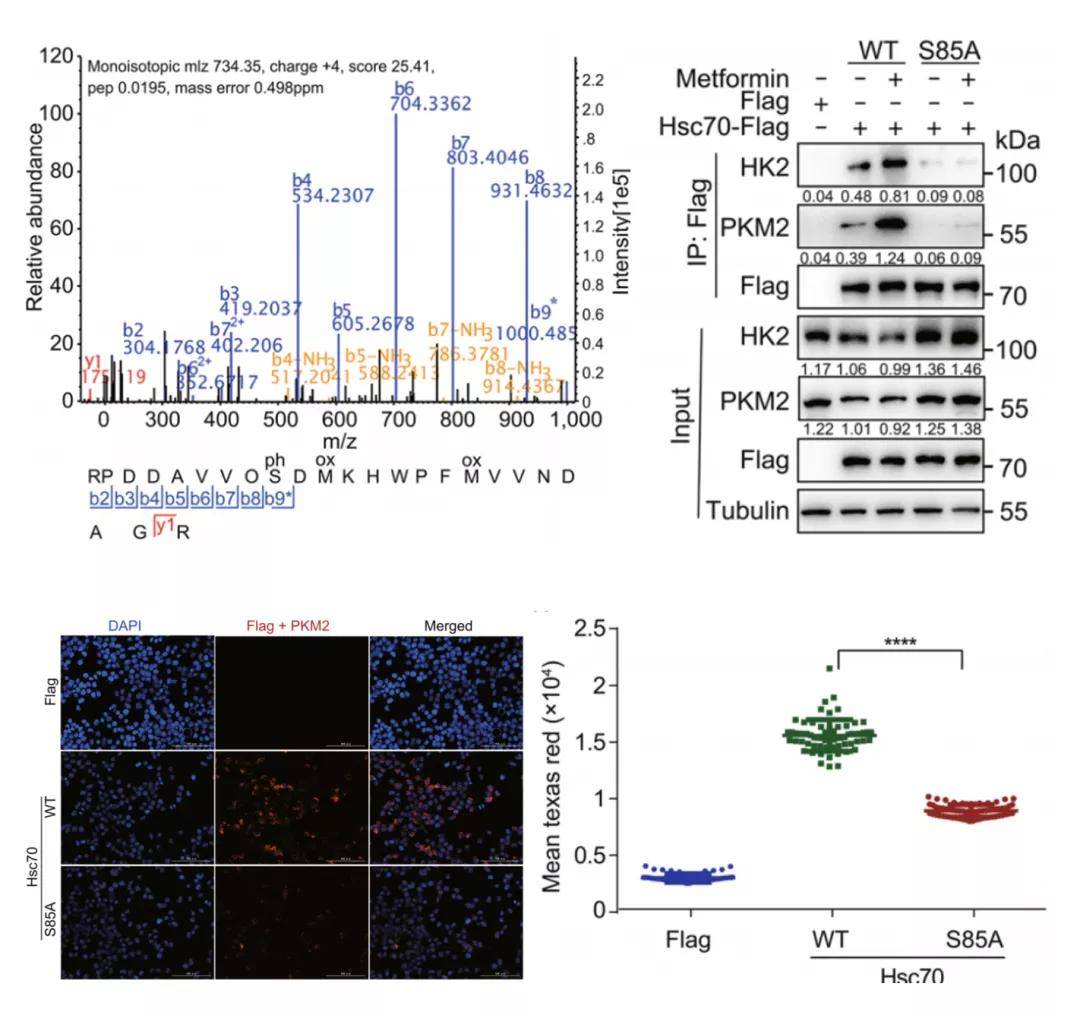
研究者发现,二甲双胍诱导的HK2-GFP荧光和蛋白水平的降低可以通过过表达λ-磷酸酶来挽救,这表明磷酸化是导致HK2-GFP降解的原因。通过对二甲双胍处理的细胞Hsc70-Flag免疫沉淀进行质谱磷酸化分析,发现二甲双胍可诱导Hsc70第85位丝氨酸发生磷酸化,其非磷酸化突变体Hsc70-S85A可显著降低二甲双胍诱导的Hsc70与CMA底物蛋白之间的相互作用。过表达Hsc70-WT而非Hsc70-S85A可降低CMA底物HK2和PKM2的蛋白水平,而敲低Lamp2a可以阻止此降解反应。这些结果表明,二甲双胍通过Hsc70 Ser85磷酸化激活CMA,促进Hsc70与CMA底物蛋白的相互作用。
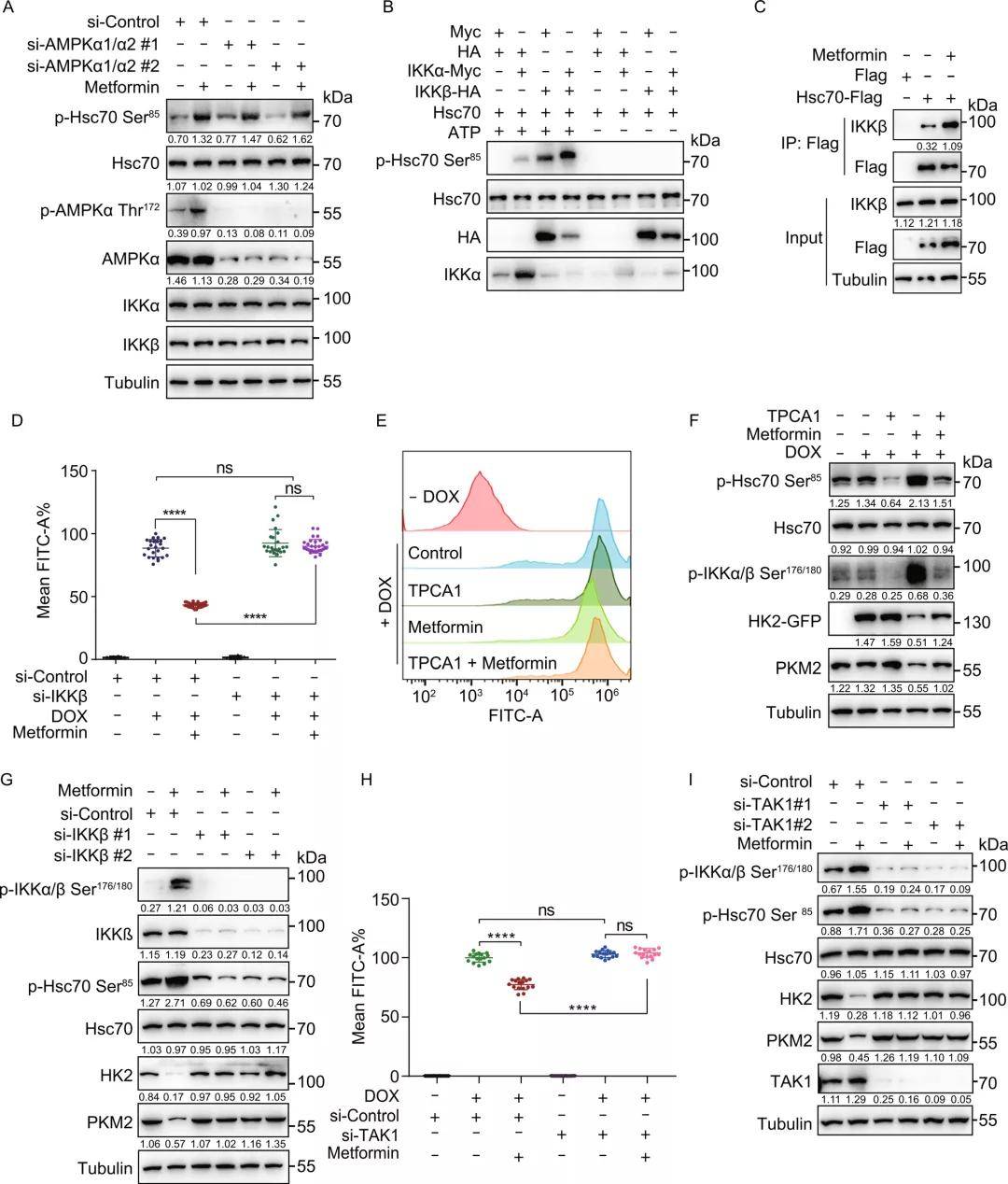
与二甲双胍诱导的CMA活化与AMPK无关一致,Hsc70 Ser85位点的磷酸化也与AMPK激活无关,研究者发现Hsc70 Ser85是IKKα/β的预测潜在靶点。体外激酶检测实验显示Hsc70的Ser85位点能被IKKα和IKKβ直接磷酸化,且IKKα和IKKβ的结合进一步加强了磷酸化。二甲双胍处理后,IKKα或IKKβ与Hsc70的相互作用增加,这表明IKKα/β在Ser85处磷酸Hsc70。敲低293THK细胞中的IKKα和IKKβ再用二甲双胍处理,发现IKKα/β敲低可以降低p-Hsc70 Ser85水平并阻止内源性HK2和PKM2的降解,挽救二甲双胍诱导的HK2-GFP荧光和蛋白质水平的降低,二者过表达则增加了p-Hsc70 Ser85水平,这些结果进一步证明,Hsc70 Ser85磷酸化的上游激酶是IKKα/β。
已知TAK1(转化生长因子β激活激酶1)能磷酸化并激活IKKα/β激酶,由此研究者验证了二甲双胍诱导的IKK和CMA激活是否依赖于TAK1。研究者发现,下调TAK1挽救了二甲双胍诱导的HK2-GFP水平下降、内源性HK2和PKM2的降解以及IKKα/β、Hsc70 Ser85的磷酸化,这说明二甲双胍可激活TAK1,进而磷酸化并激活IKKα/β, IKKα/β再磷酸化Hsc70诱导CMA的激活。体外热位移法检测证明二甲双胍与TAK1之间没有直接的相互作用,即TAK1不是二甲双胍的直接靶点,但它参与了二甲双胍诱导的CMA激活。
随后,研究者研究了二甲双胍激活CMA后APP是否可以被靶向降解,发现甲双胍以溶酶体、Hsc70和LAMP2A依赖的方式诱导SH-SY5Y细胞内源性APP蛋白的降解。同时,也增强了内源APP与Hsc70-Flag以及LAMP2A-Flag之间的相互作用,且Hsc70-Flag与APP的相互作用能被Hsc70的S85A突变所阻断。值得注意的是,抑制IKKβ和TAK1的表达可以阻断二甲双胍诱导的APP降解,同时阻断Hsc70(Ser85)和IKKα/β(Ser176/180)的磷酸化。这些结果表明,APP是CMA的底物,二甲双胍可以通过激活TAK1-IKKα/β-Hsc70-CMA途径来诱导其降解。

经过探究研究者发现,二甲双胍诱导的CMA激活可以诱导APP降解并减轻APP和Aβ的细胞毒性作用。接着,研究者又对APP/PS1小鼠进行了行为学实验,探索了二甲双胍诱导的CMA活化对小鼠记忆丧失及AD相关分子标志物的影响,发现二甲双胍处理后,APP/PS1小鼠的学习和空间记忆得到了明显改善,整个大脑中不溶性Aβ1-42水平显著下降,海马中Aβ斑块水平下降。GFAP染色发现,二甲双胍处理后海马星形胶质细胞的激活也有所降低。此外,与体外实验结果一致,二甲双胍处理也显著降低了APP蛋白水平,并诱导了Hsc70 Ser85位点的磷酸化。
为验证Hsc70过表达激活CMA是否也可以缓解APP/PS1表型,研究者利用AAV在小鼠的海马区对Hsc70 WT和Hsc70-S85A进行了过表达。发现Hsc70 WT过表达减轻了APP/PS1小鼠的认知缺陷,并且有效降低了APP/PS1小鼠脑组织中不溶性Aβ1-42水平及其海马区Aβ斑块水平和APP蛋白水平。这一结果表明,通过二甲双胍或过表达Hsc70激活CMA可降低不溶性Aβ1-42积累、Aβ斑块水平,诱导APP降解,从而缓解APP/PS1小鼠的认知缺陷。
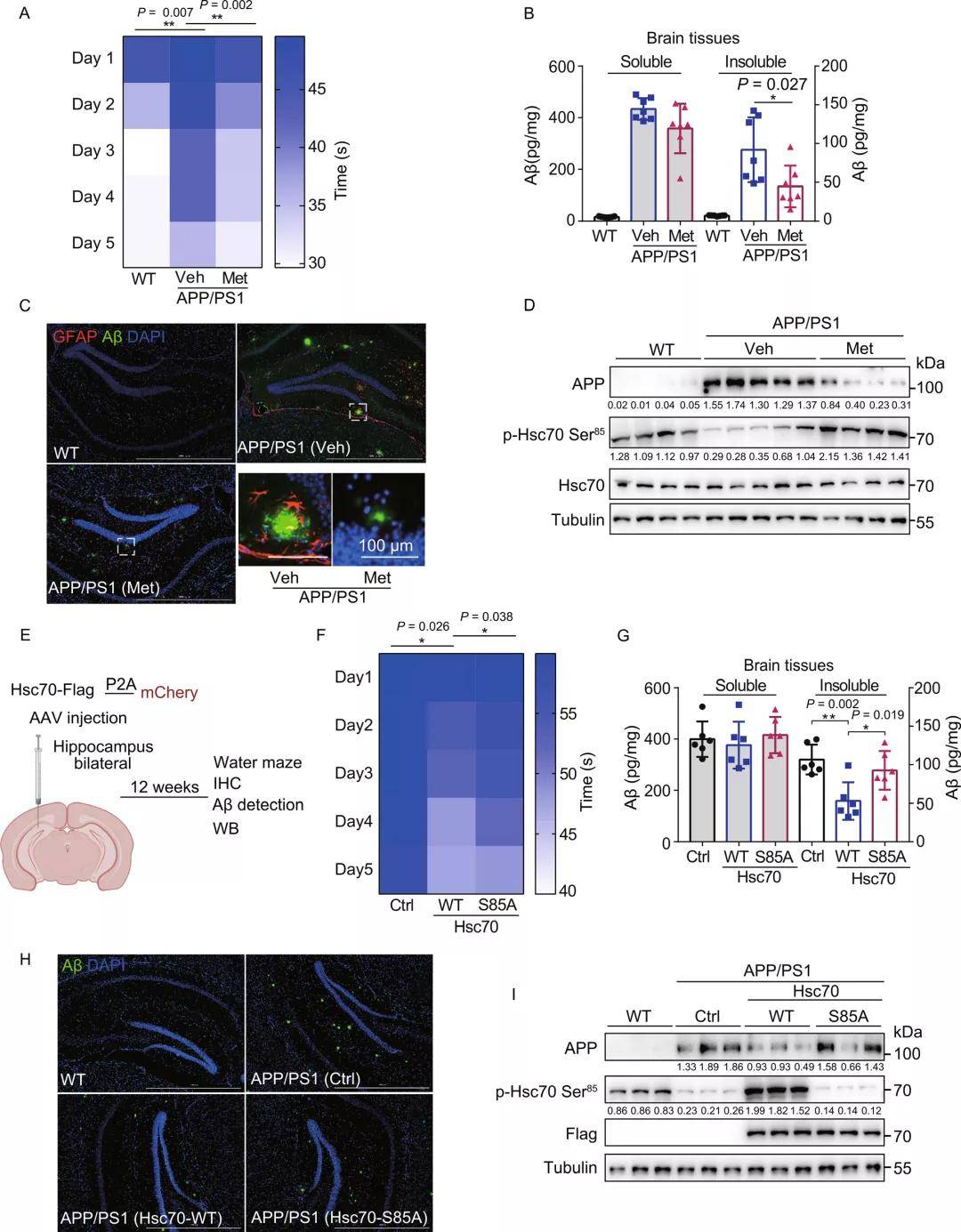
综上所述,该研究发现降糖药物二甲双胍可作为CMA的激活剂,同时也证实了APP是CMA的底物,二甲双胍可以通过激活TAK1-IKKα/β-Hsc70-CMA途径来诱导其降解,进而改善AD小鼠的认知缺陷;除了作为治疗2型糖尿病的临床药物,二甲双胍也初次被证实具有用于治疗AD疾病的可行性。总之,该研究揭示了二甲双胍通过激活分子伴侣介导自噬治疗 AD 的新机制,为AD等相关退行性神经疾病的治疗提供了一种潜在的治疗策略。
(点击“阅读原文”查看原文内容)

400-077-2566

service@wzbio.cn







