

 当前位置:首页 > 研究领域 > 循环系统
当前位置:首页 > 研究领域 > 循环系统
心肌肥厚是心力衰竭的主要原因之一,但有效的治疗靶点仍然有限,而跨膜通道样蛋白6(TMC6)在健康心肌中含量丰富,但在肥厚心肌中表达下调,其在心肌肥厚中的作用尚不清楚。浙江大学医学院附属第一医院梁平教授团队联合复旦大学脑科学研究院唐逸泉教授团队在Circulation Research(IF 16.2)发表题为“TMC6 Is a Novel Therapeutic Target for Pathogenic Cardiac Hypertrophy”的文章。研究揭示TMC6是病理性心肌肥厚的内源性抑制因子,通过将CIB1隔离在内质网中,从而抑制CIB1-钙调神经磷酸酶/NFAT信号通路,提示TMC6–CIB1轴是一个潜在的治疗靶点。

| 基因信息 | TMC6:跨膜通道样蛋白6 |
|---|---|
| 实验动物 | 8周龄WT和 Tmc6 敲除小鼠 |
| 病毒产品 | AAV9-cTNT-TMC6;AAV9-cTNT-GFP |
| 注射方式 | 尾静脉注射 |
| 病毒用量 | 1×1012 genomic copies/mouse |
研究数据显示在人类与小鼠心脏中,TMC6是TMC家族中表达最高的成员,并且在三种心肌肥厚模型中,TMC6蛋白水平均显著下降。心肌特异性Tmc6敲除小鼠出现轻度肥厚,并在TAC后表现出更严重的心功能下降、肥大、纤维化和生存率降低。为了进一步研究TMC6是否可以以细胞自主的方式预防压力超负荷诱导的心脏肥大,研究团队将AAV9-cTNT-TMC6及对照病毒注射至WT和Tmc6敲除小鼠体内;载体给药后4周,小鼠接受TAC手术。与表达GFP的对照组相比,心脏特异性过表达TMC6的WT-TAC和KO-TAC小鼠显著降低了心脏肥大和功能障碍,体现TMC6在防止TAC诱导的心肌肥大中的细胞自主作用。进一步的RNA-Seq分析表明Tmc6基因敲除通过激活特定的TF,如SRF,MEF家族和NFAT家族,加剧心肌肥厚。
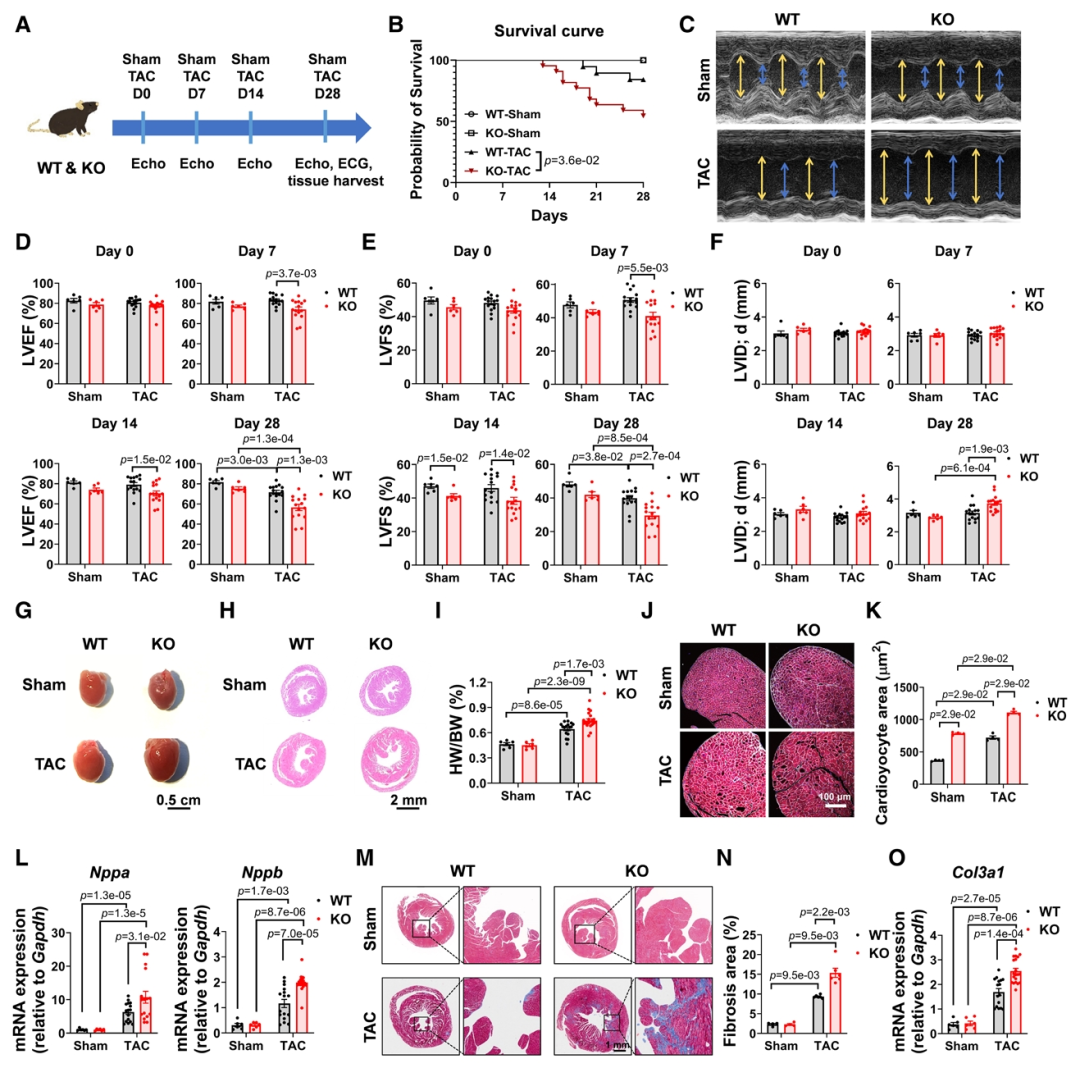
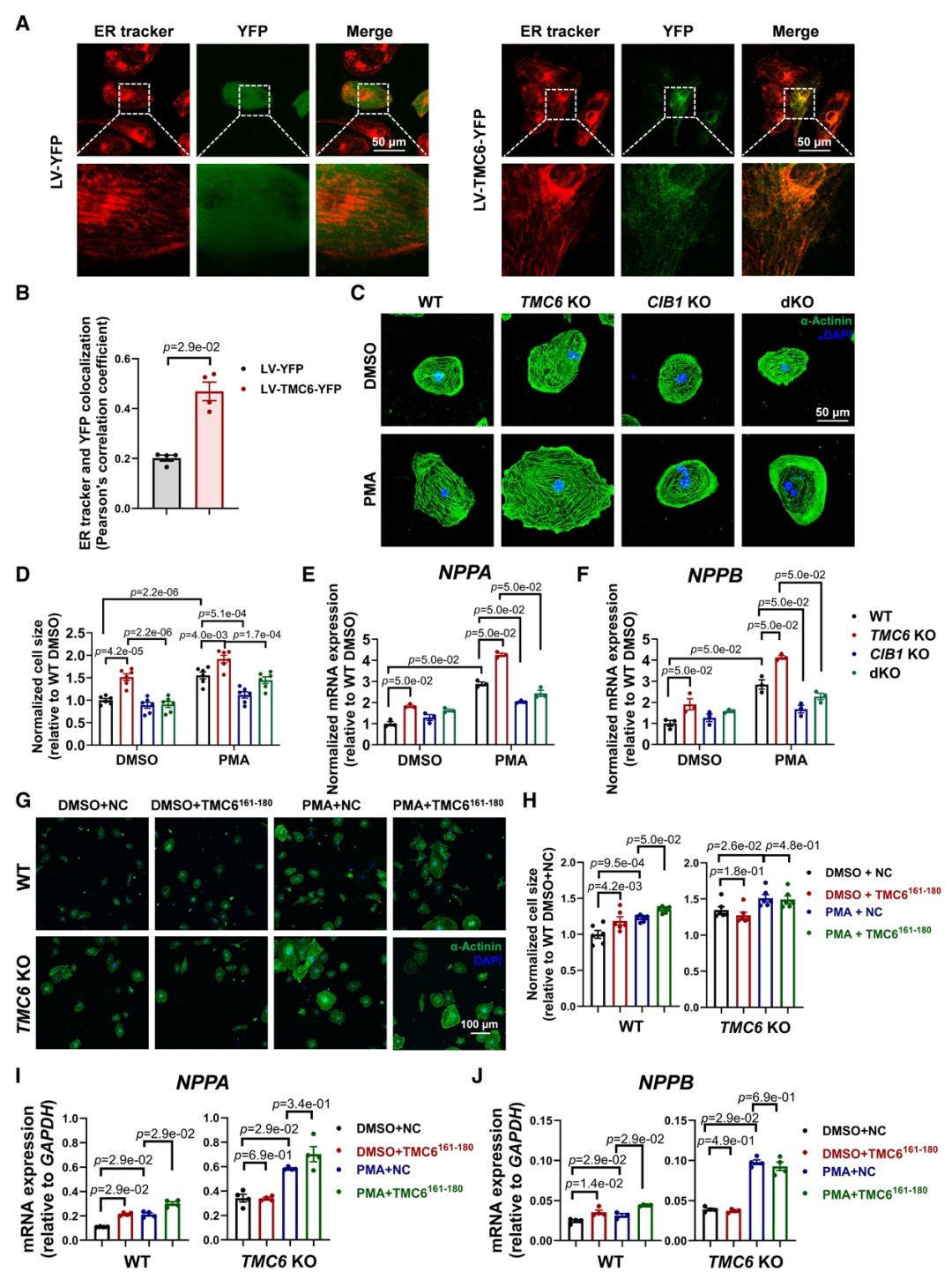
体外TMC6敲低及敲除的细胞在肥厚刺激下,钙调神经磷酸酶活性升高、NFATc4 核转位增加,肥厚标志物表达上调,该效应可被钙调神经磷酸酶抑制剂逆转,表明TMC6缺乏可通过激活钙调磷酸酶/NFAT信号通路导致心肌细胞肥大。通过酵母双杂交筛选及免疫共沉淀等分析,证实TMC6与CIB1存在相互作用,TMC6的161-180氨基酸片段为CIB1结合域,该片段可竞争性破坏TMC6-CIB1结合。亚细胞定位实验发现TMC6定位于内质网(ER),当CIB1单独表达时,主要定位于质膜和细胞核,在与TMC6共表达时,CIB1则重新定位于ER;此外竞争性肽TMC6161-180的共表达可阻断这种重新定位。实验进一步证实敲除CIB1能够逆转因TMC6缺失所诱发的心肌肥厚表型;TMC6161-180将CIB1从TMC6上置换下来,从而增加了游离CIB1的池容量,使其能够在肌膜微区招募钙调神经磷酸酶并激活NFAT依赖的促肥厚信号通路。
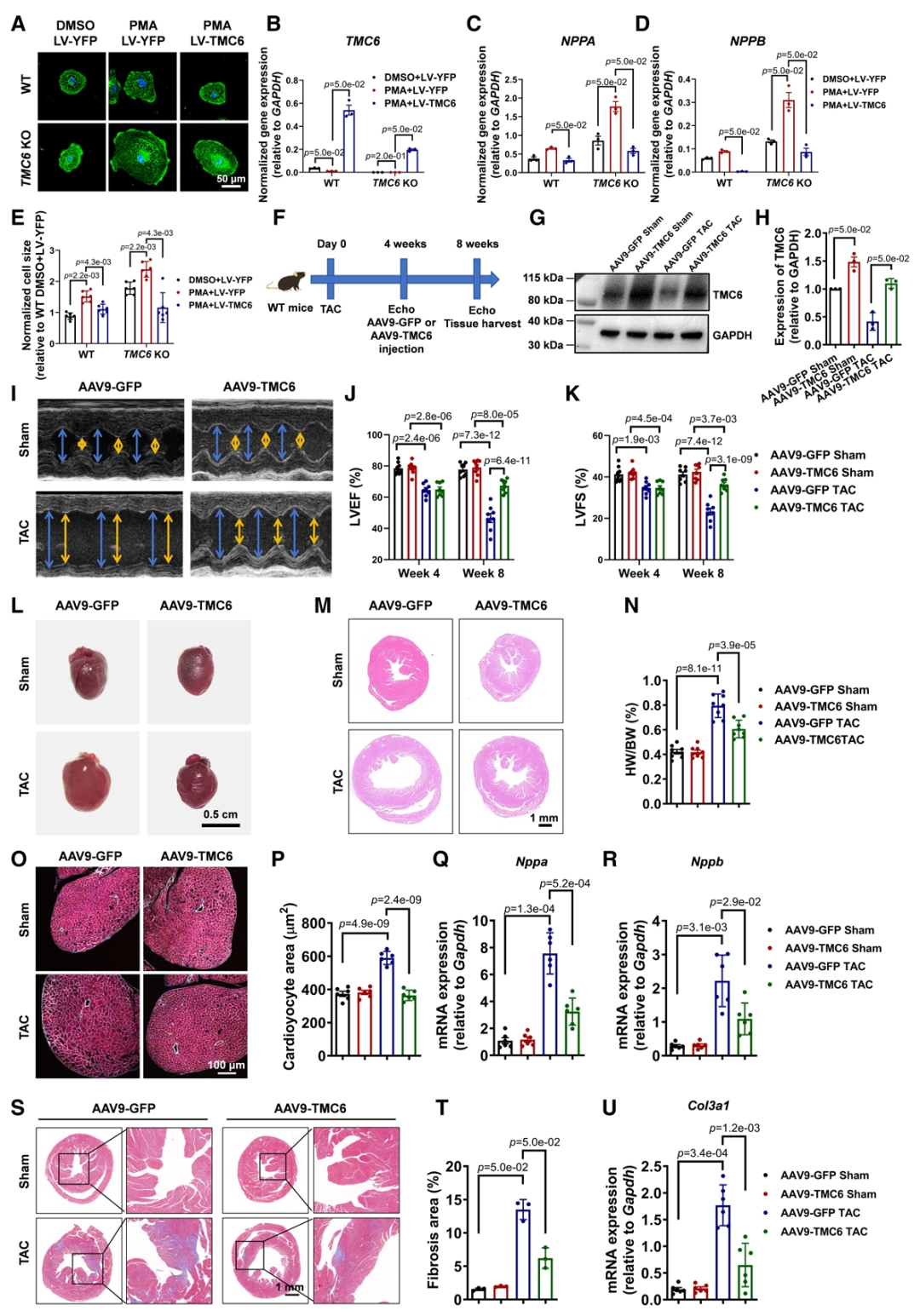
基于上述的结果,研究团队提出假设:TMC6可能是心肌肥厚的一个治疗靶点,随之进行了体外和体内研究。体外过表达TMC6可下调PMA诱导的肥大基因表达,并使细胞体积缩小。为了研究TMC6基因治疗对体内心脏肥大的治疗效果,WT小鼠在假手术或TAC手术后4周接受尾静脉注射AAV9-cTNT-TMC6或AAV9-cTNT-GFP。评估结果显示TMC6过表达显著恢复了TMC6蛋白水平,改善了心脏功能,减轻了心肌细胞的肥大以及心脏纤维化。这些结果表明,TMC6基因治疗有效地减轻体外和体内模型的病理性心脏肥大。
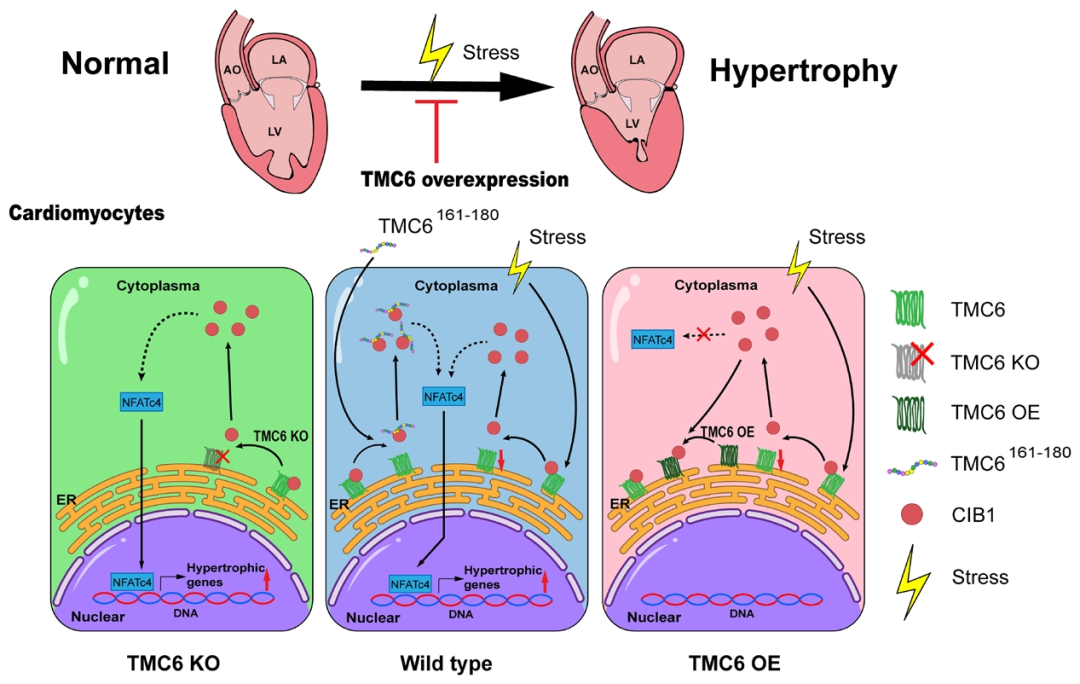
研究揭示了TMC6在心肌肥厚中的新作用,证明其通过CIB1-钙调神经磷酸酶-NFAT信号通路发挥抗肥厚效应。这些发现表明,TMC6是治疗病理性心肌肥厚和心力衰竭的潜在治疗靶点。

400-077-2566

service@wzbio.cn







