

 当前位置:首页 > 研究领域 > 循环系统
当前位置:首页 > 研究领域 > 循环系统
通过上一期AAV在心脏研究中的靶向策略(干货篇)对rAAV在心脏实验中的启动子、血清型以及注射方式的选择策略的学习,相信大家对rAAV在心脏研究中的应用已有所了解,今天小V将和大家一起,通过几个案例深入了解一下关于AAV在心脏研究中的具体应用。
ZBTB20(锌指和BTB结构域蛋白20)是来自POK家族的转录因子,被证明在维持心脏收缩中发挥关键作用,但在心脏重构中的作用尚未被阐明。本研究中,作者用血管紧张素II(Ang II)诱导小鼠心脏重塑模型,探讨ZBTB20在心脏重构中的作用。研究发现ZBTB20在Ang II诱导的心脏重塑和心肌细胞损伤反应中表达升高;AAV9(心肌注射)介导的ZBTB20过表达,可导致心壁肥厚、心室扩张、纤维化增加和射血分数降低,而敲低ZBTB20后可保护心肌细胞免受Ang II诱导的肥大影响。从分子机制来看,ZBTB20通过EGFR-Akt信号通路加重Ang II诱导的心脏重塑反应,本研究阐明了ZBTB20在不良心脏重构向心力衰竭的转变中的作用,并为诱导不良心脏重构的分子机制提供了依据。
| 病毒产品 | AAV9-ZBTB20 & AAV9-shZBTB20 |
|---|---|
| 实验动物 | 8-10周龄雄性C57BL/6小鼠 |
| 注射方式 | 心肌注射 |
| 注射量 | 10 μl/site,1*10E11vp,三点注射 |
如图1所示,使用AAV9病毒载体在小鼠心脏中过表达ZBTB20, ZBTB20-NS组和ZBTB20-Ang II组ZBTB20蛋白表达水平均明显升高,并且ZBTB20-Ang II组的ZBTB20蛋白表达水平高于ZBTB20-NS组。Ang II处理四周后,ZBTB20注射组较NC组心肺重量更高,同时小鼠左心室和心肌细胞肥大增加,这提示ZBTB20组出现心肌肥大和肺水肿。此外,多种不良心脏重构和纤维化分子标志物转录增加,血管周围和间质纤维化变得更为严重,这些数据表明ZBTB20的过表达促进了不良的心脏重构发展为心力衰竭。
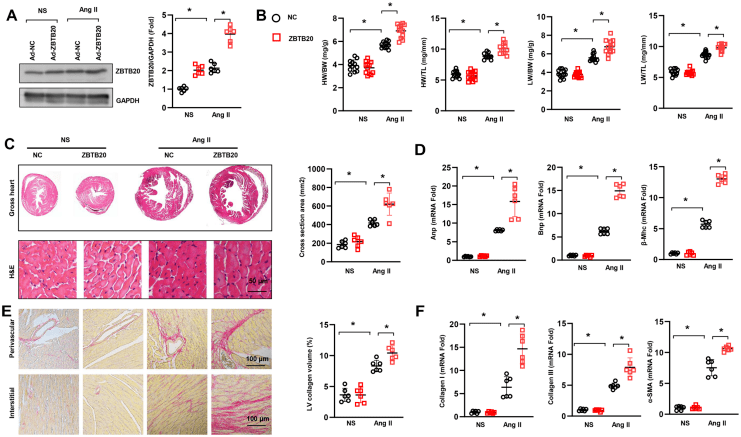
如图2所示,使用AAV9 RNA干扰ZBTB20,WB检测到ZBTB20蛋白水平大大降低。小鼠心肌细胞经转染ZBTB20 siRNA后肥大反应明显降低,而且细胞表面积变小,心脏重构分子标志物Anp和β-Mhc的转录水平也有所降低,表明敲低ZBTB20可保护心肌细胞免受Ang II诱导的肥大影响。
由各种心脏疾病导致的心力衰竭(HF)是造成当今社会人群死亡的主要原因之一,目前尚无特别有效的治疗方法,寻找新的标志物对HF的诊断和治疗具有重要意义。前期研究发现miR-590-3p可显著增加幼鼠及成年大鼠心肌细胞的增殖,并且在肺动脉高压儿童患者上腔静脉血液中表达上调。网状内皮素4(RTN4)被证实在肥厚性心肌病动物模型中显著上调,同时参与冠心病调控,可作为心衰的指标。此外,RTN4还是miR-590-5p的预测下游靶点。基于这些研究结果,研究人员探索了miR-590-3p和RTN4在HF中的关系。
研究人员采用主动脉弓缩窄术制备小鼠HF模型,并对小鼠心力衰竭的的病理进程进行分析观察,发现miR-590-5p在HF小鼠心脏组织中表达下调,尾静脉注射AAV9-miR-590-5p可减轻心肌肥厚和心肌细胞凋亡。此外,miR-590-5p过表达可促进H9C2细胞活力,抑制细胞凋亡,降低心肌重塑水平。机制上,miR-590-5p结合到RTN4 3 'UTR,通过负调控RTN4 调节心肌细胞表型。综上所述,miR-590-5p通过下调RTN4调控HF心肌肥厚和心肌细胞凋亡。
| 病毒产品 | AAV9-miR-590-5p & AAV9-NC |
|---|---|
| 实验动物 | 8-10周龄雄性C57BL/6小鼠 |
| 注射方式 | 尾静脉注射 |
| 病毒滴度 | 1*10E12vg/mL |
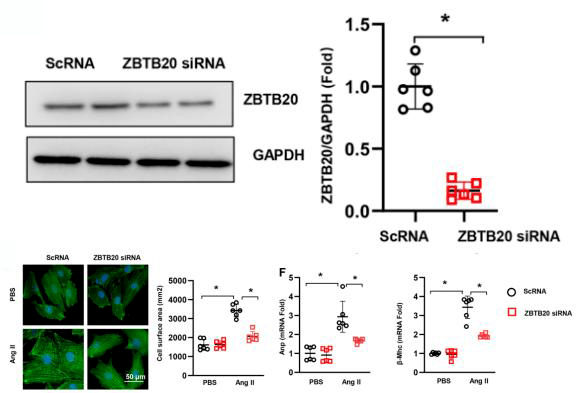
如图3所示,通过RT-qPCR验证AAV9-miR-590-5p在小鼠中的表达大幅提高。miR-590-5p的过表达降低了小鼠LVEDD(左心室舒张末期内径),减轻了小鼠心肌的水肿和肥厚。此外,miR-590-5p的过表达还显著降低了小鼠心脏组织ANF、BNP和β-MHC(心脏重构标志物)的蛋白水平,同时使Bcl-2(细胞凋亡抑制基因)蛋白水平升高及Bax(细胞凋亡促进基因)蛋白水平降低。综上所述,miR-590-5p过表达可减轻HF模型小鼠的心肌肥厚和心功能障碍。
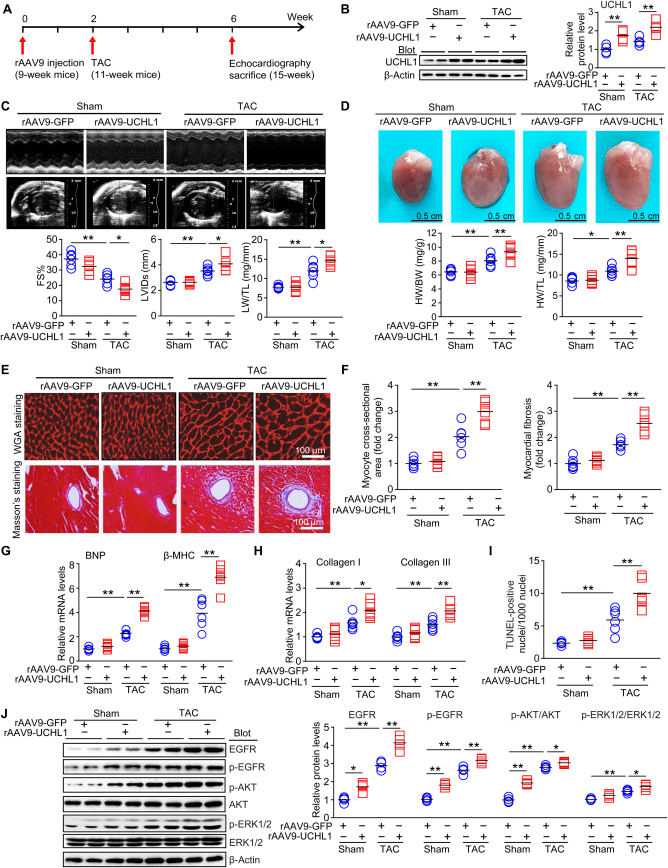
病理性心脏肥大可导致心力衰竭。研究证实在促心肌肥厚途径中表皮生长因子受体(EGFR或ErbB1)是正调控因子,EGFR的稳定性、转运和激活主要受翻译后修饰的调控,然而其具体调控机制尚不完全清楚。研究表明去泛素化酶UCHL1在心肌梗死后的心肌细胞中表达高度上调,但是人们对其在调节心肌肥厚和重构中的作用知之甚少。
在本研究中,研究者通过功能缺失和功能获得的方法并使用抑制剂,证实UCHL1在激动剂诱导的心肌肥厚细胞及衰竭心脏中表达上调,过表达UCHL1(AAV9,尾静脉注射)可加速压力超负荷诱导的心肌肥厚,而下调UCHL1则可显著改善由激动剂或压力超负荷引起的心肌肥厚;同时阐明了UCHL1通过降低EGFR泛素化和降解正向调节心肌肥厚,全身给药UCHL1抑制剂可显著逆转心肌肥厚和重构。综上所述,这些发现表明UCHL1在心肌肥厚的发病机制中发挥重要作用,可作为肥厚治疗的靶点。
| 病毒产品 | rAAV9-CMV-UCHL1 & rAAV9-CMV-GFP |
|---|---|
| 实验动物 | 9周龄C57BL/6J小鼠 |
| 注射方式 | 尾静脉注射 |
| 注射量 | 5*10E11vg |
| 病毒滴度 | 1*10E13 - 3.5*10E13 vg/mL |
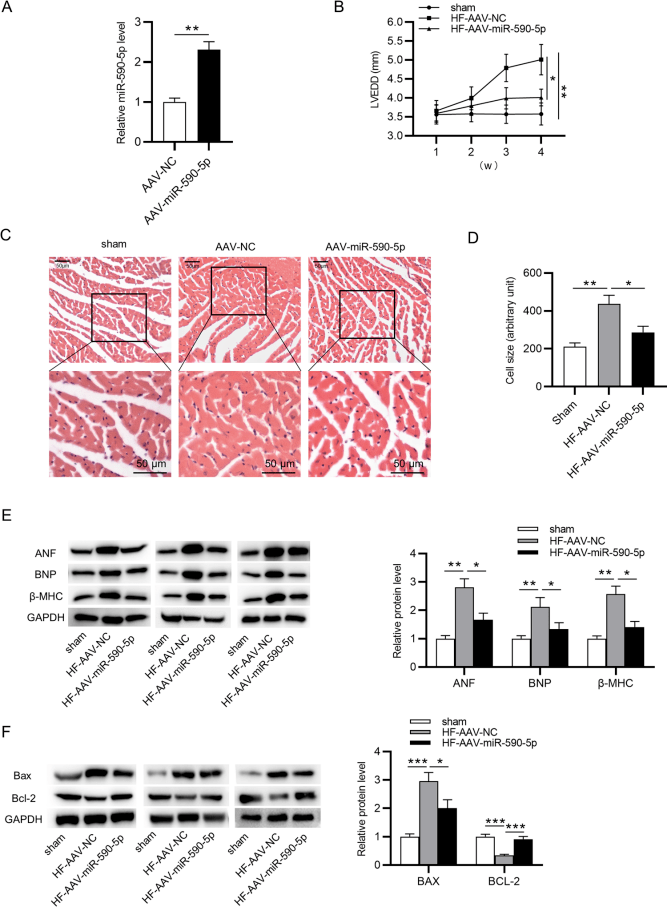
如图4所示,利用AAV9载体在小鼠心脏中过表达UCHL1,荧光显微镜检测显示心脏细胞得到高效感染,免疫印迹分析显示心脏中UCHL1蛋白水平大大提高。
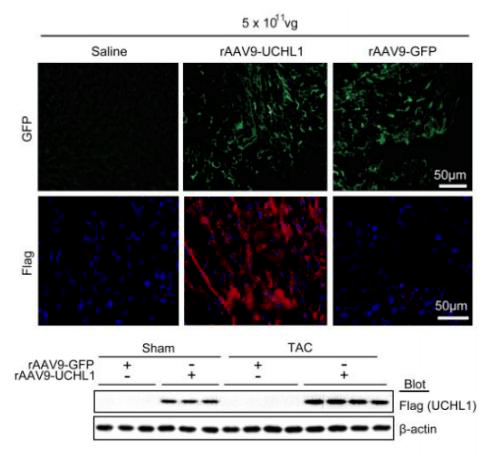
与rAAV9-GFP对照组相比,UCHL1的过表达导致TAC术后小鼠发生更严重的心功能障碍,且死亡率明显增加。此外在UCHL1表达增加的同时,EGFR蛋白水平、小鼠心肌中磷酸化的EGFR、AKT和ERK1/2(促心肌肥厚因子)也显著升高,表明心脏中过表达UCHL1可加重心肌肥厚,并可能与EGFR的稳定性有关(图5)。
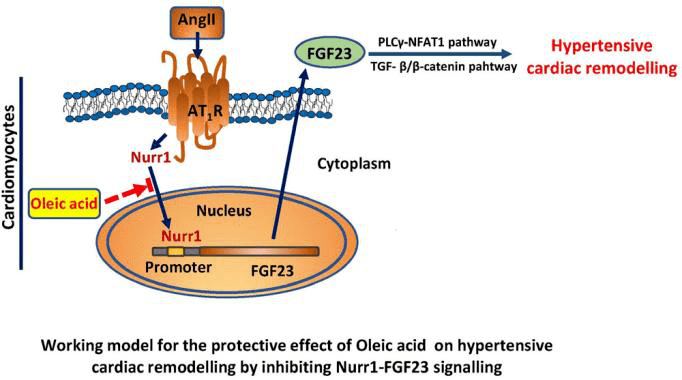
高血压可损伤多种靶器官,研究表明在轻度至中度高血压人群中左心室肥厚(LVH)患病率为20%至50%。然而,高血压心脏重构的病理机制仍知之甚少。油酸(OA)是一种重要的单不饱和脂肪酸,据报道富含OA的地中海饮食可降低心血管在内的多种疾病的发生率,但其在高血压性心脏重塑中的作用及机制尚不清楚。
本研究中作者对OA在高血压性心脏重塑中的作用进行了探讨。通过检测伴有和不伴有LVH高血压患者的血浆代谢谱,研究人员发现伴LVH高血压患者的外周血中OA水平显著降低,并先后通过体外及体内研究证实OA可显著降低Ang II(血管紧张素II)诱导的心肌细胞生长、心肌成纤维细胞胶原表达和小鼠心脏重塑。RNA测序进一步发现FGF23(成纤维细胞生长因子23)在Ang II诱导反应中表达显著上调,而在OA处理后表达明显受抑。利用AAV9载体过表达FGF23可加重Ang II诱导的心脏重塑,并削弱OA对心脏重塑的保护作用,进一步研究发现OA通过阻断Nurr1(核受体相关蛋白)从细胞质到细胞核的易位抑制Ang II诱导的FGF23表达,深入揭示了OA在防止Ang II诱导的心脏重塑中发挥关键作用。
| 病毒产品 | AAV9-FGF23 & AAV9-GFP |
|---|---|
| 实验动物 | 10周龄C57BL/6雄性小鼠 |
| 注射方式 | 尾静脉注射 |
| 注射量 | 150μl;5*10E11VG/ml(稀释后) |
| 病毒滴度 | 5.94*10E13VG/ml & 1.5*10E14VG/ml |
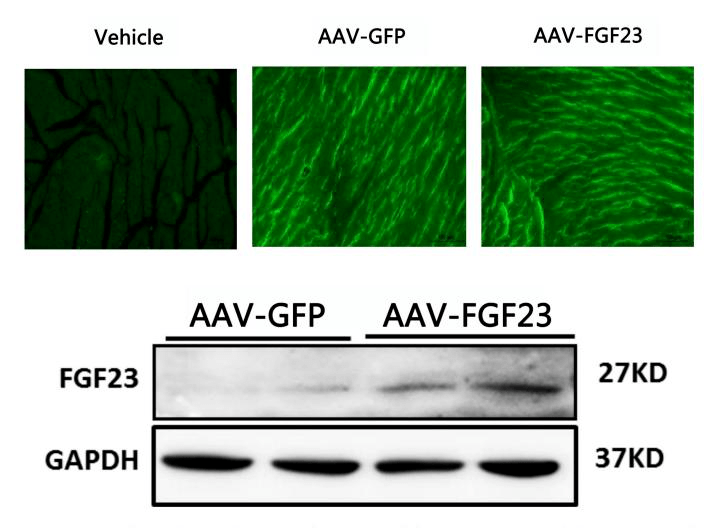
通过尾静脉注射将AAV9-FGF23及AAV9-GFP载体注入小鼠体内,荧光显微镜及WB检测显示AAV9病毒高效传递至小鼠心脏,FGF23蛋白在心脏组织中成功过表达(图6)。FGF23在心脏组织中的过表达加重了Ang II诱导的心功能障碍,并损害了OA对心脏重构及心肌纤维化的保护作用。
心肌梗死(MI)是全球发病率和死亡率最高的心脏疾病之一,梗死后会发生心肌重塑并可能导致心力衰竭。目前,药物干预仍是预防梗死后心脏重塑和功能障碍的有效治疗方法,因此,寻找有效的干预靶点,减轻心肌梗死后心脏重构,从而防止梗死后患者心力衰竭的发展是至关重要的。据报道,细胞凋亡和自噬参与心肌组织发生的生理过程,而且哺乳动物雷帕霉素靶蛋白(mTOR)被证实在许多心脏病中可同时调节凋亡和自噬相关的发病机制,但其具体调控机制尚不清楚。发育及DNA损伤反应调节基因1(Redd1)参与mTOR活性调节,但其在心脏中的作用知之甚少。
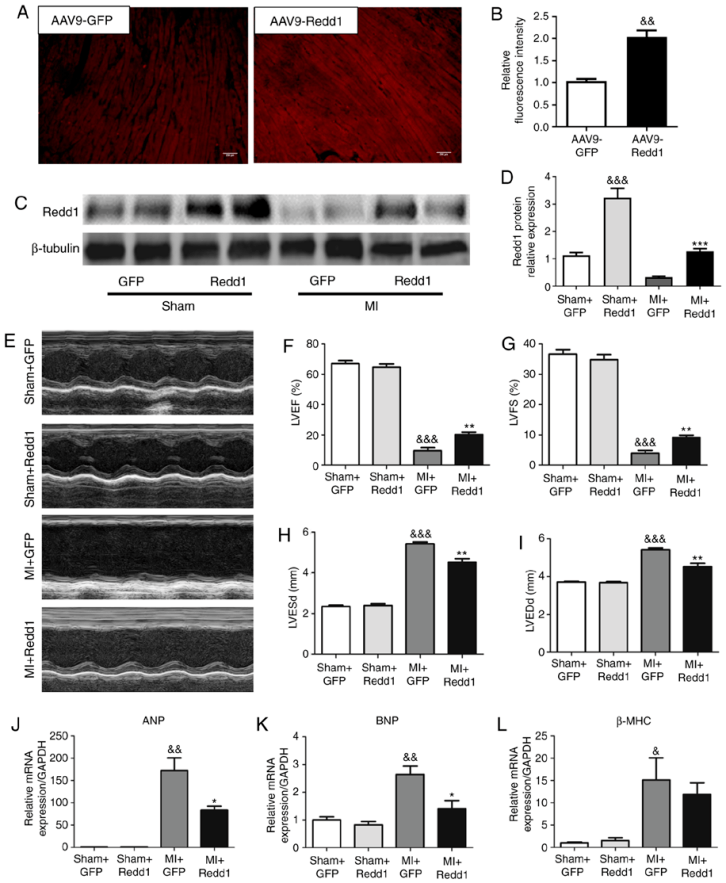
在本研究中,作者首先检测了MI小鼠心脏中Redd1的表达情况,发现Redd1表达量下调。进一步使用AAV9介导Redd1过表达发现Redd1的过表达改善了小鼠左心室功能障碍,降低了扩张指数,并抑制了心肌细胞凋亡及改善了自噬。此外,研究发现过表达Redd1可以抑制mTOR及其下游效应蛋白的磷酸化,进而缓解缺血损伤引起的心室功能障碍。综上所述,本研究证明Redd1过表达通过mTOR信号通路减少细胞凋亡和增强自噬来保护心肌梗死后心力衰竭的发生和持续,深入揭示了Redd1是心肌梗死后心力衰竭发展的治疗靶点。
| 病毒产品 | AAV9-Redd1 & AAV9-GFP |
|---|---|
| 实验动物 | 4-5周龄C57BL/6雄性小鼠 |
| 注射方式 | 尾静脉注射 |
| 注射量 | 2.8*10E11VG/mice |
研究人员通过尾静脉注射AAV9载体上调Redd1的表达,发现小鼠心肌细胞中Redd1表达量显著上升。Redd1的过表达显著降低了LVEDd和LVESd,大大提高了LVEF 和LVFS水平,并使ANP和BNP mRNA水平大幅下降,同时β-MHC mRNA水平也略有下降。总的来说,Redd1过表达在心肌梗死后对心功能障碍和心脏扩张有保护作用。
舒尼替尼(SNT)等酪氨酸激酶抑制剂可以显著提高癌症患者的预期寿命,但是这些药物的使用可能会引发心脏收缩功能障碍、高血压、慢性心力衰竭等心血管方面的不良反应。然而,这种作用的分子机制仍然不清楚。MicroRNA是心血管疾病和酪氨酸激酶途径中的关键调控因子,已被证实在心血管疾病中发挥着关键作用。MicroRNA的过表达和下调可能是治疗心脏病患者的一种有效方法。
研究人员使用miRNA芯片分析了舒尼替尼处理后小鼠心肌中miRNA的差异表达,发现miR-146a显著下调。利用荧光素酶报告基因检测实验证实miR-146a的下游靶点为Pln和Ank2,二者与心脏收缩功能障碍密切相关。体内外实验结果表明SNT下调miR-146a的表达,并通过调控下游靶点PLN和ANK2导致心脏收缩功能障碍,上调miR-146a可缓解SNT对心脏收缩力的抑制作用。本研究揭示了miR-146a可能是一种有效的SNT诱导的心脏功能障碍的保护剂。
| 病毒产品 | AAV9-cTNT-GFP-miR-146a & AAV9-Control |
|---|---|
| 实验动物 | C57雄性小鼠(20-25g) |
| 注射方式 | 尾静脉注射 |
| 病毒滴度 | 1*10E11VP |
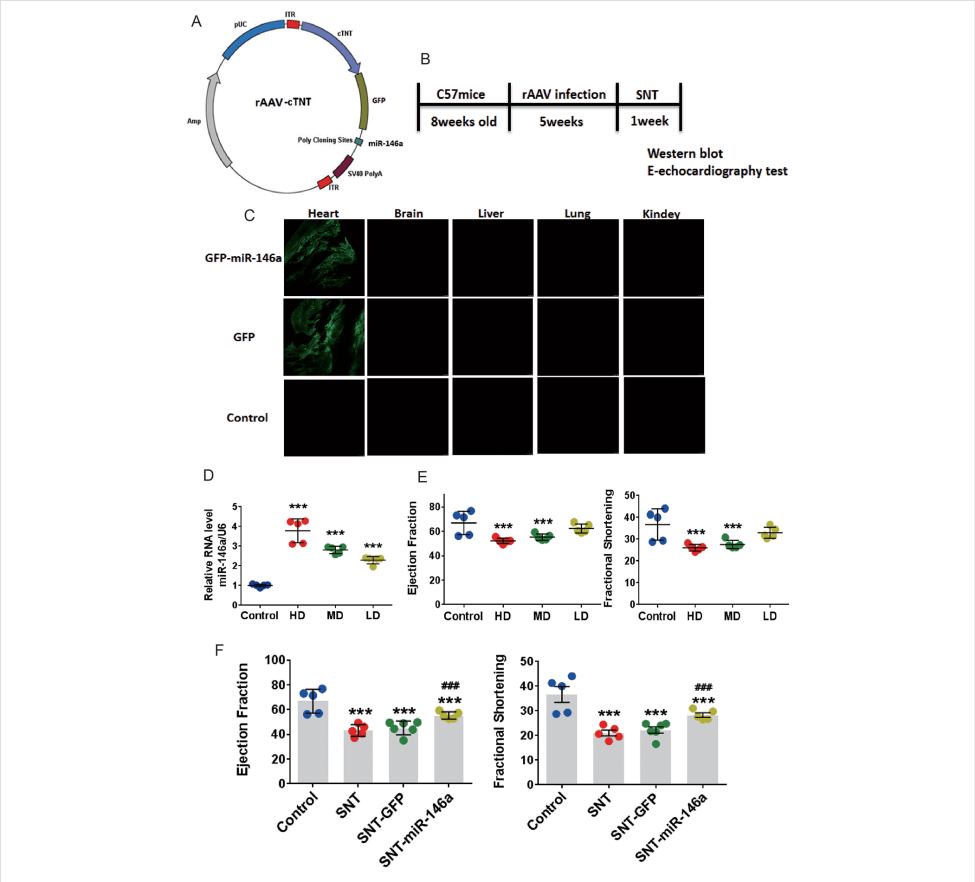
如图8所示,研究人员使用心脏特异性启动子cTNT驱动的AAV9载体在小鼠心脏特异表达miR-146a,不同组织的荧光图像表明,静脉注射病毒载体后仅心脏被高效感染。通过超声心动图检测心脏收缩功能,结果表明心脏中miR-146a的过表达显著抑制了SNT诱导的EF和FS降低,表明心脏功能得到改善。

400-077-2566

service@wzbio.cn







